Molecular basis and genetic testing strategies for diagnosing 21-hydroxylase deficiency, including CAH-X syndrome
- Affiliations
-
- 1Department of Pediatrics, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea
- 2Medical Genetics Center, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea
- 3Department of Pediatrics, Bundang CHA Medical Center, CHA University, Seongnam, Korea
- KMID: 2543288
- DOI: http://doi.org/10.6065/apem.2346108.054
Abstract
- Congenital adrenal hyperplasia (CAH) is a group of autosomally recessive disorders that result from impaired synthesis of glucocorticoid and mineralocorticoid. Most cases (~95%) are caused by mutations in the CYP21A2 gene, which encodes steroid 21-hydroxylase. CAH patients manifest a wide phenotypic spectrum according to their degree of residual enzyme activity. CYP21A2 and its pseudogene (CYP21A1P) are located 30 kb apart in the 6q21.3 region and share approximately 98% of their sequences in the coding region. Both genes are aligned in tandem with the C4, SKT19, and TNX genes, forming 2 segments of the RCCX modules that are arranged as STK19-C4A-CYP21A1P-TNXA-STK19B-C4B-CYP21A2-TNXB. The high sequence homology between the active gene and pseudogene leads to frequent microconversions and large rearrangements through intergenic recombination. The TNXB gene encodes an extracellular matrix glycoprotein, tenascin-X (TNX), and defects in TNXB cause Ehlers-Danlos syndrome. Deletions affecting both CYP21A2 and TNXB result in a contiguous gene deletion syndrome known as CAH-X syndrome. Because of the high homology between CYP21A2 and CYP21A1P, genetic testing for CAH should include an evaluation of copy number variations, as well as Sanger sequencing. Although it poses challenges for genetic testing, a large number of mutations and their associated phenotypes have been identified, which has helped to establish genotype-phenotype correlations. The genotype is helpful for guiding early treatment, predicting the clinical phenotype and prognosis, and providing genetic counseling. In particular, it can help ensure proper management of the potential complications of CAH-X syndrome, such as musculoskeletal and cardiac defects. This review focuses on the molecular pathophysiology and genetic diagnosis of 21-hydroxylase deficiency and highlights genetic testing strategies for CAH-X syndrome.
Figure
-
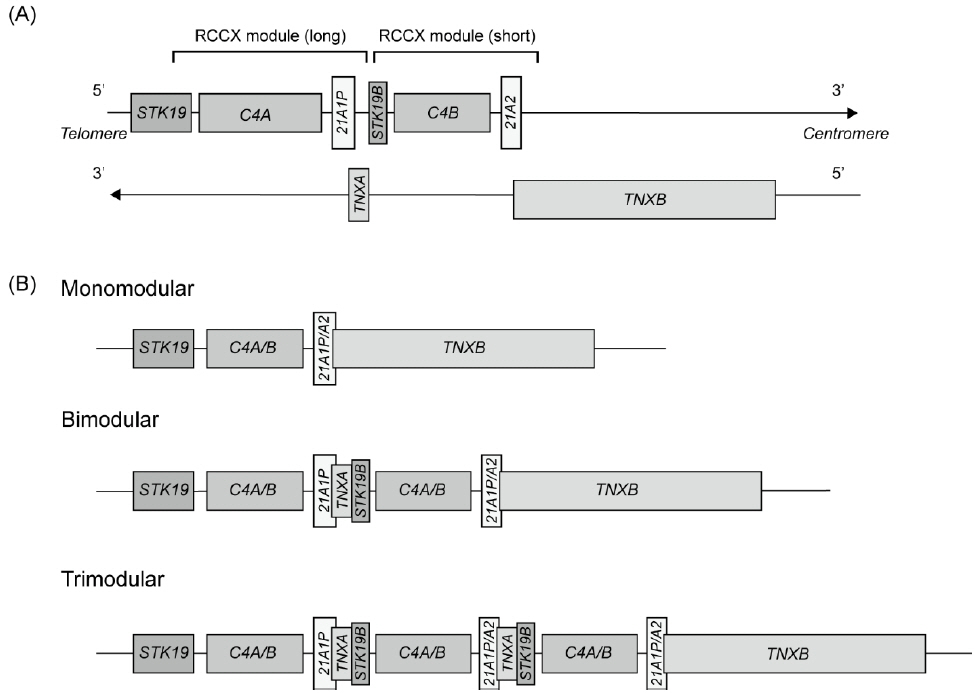
Fig. 1. Organization of the human RCCX module and copy number variations on chromosome 6p21. (A) Schematic representation of the RCCX module featuring 2 segments with the following gene arrangement: STK19 (formerly RP1)-C4A-CYP21A1P-TNXA-STK19B-C4B-CYP21A2-TNXB. The arrow indicates transcriptional orientation. (B) Three different RCCX module arrangements: monomodular, bimodular, and trimodular structures.
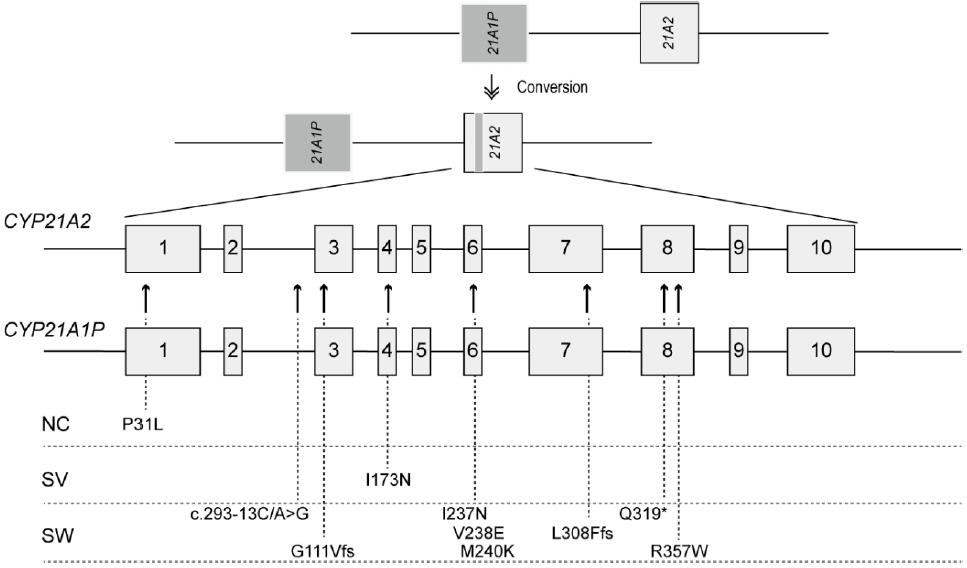
Fig. 2. Schematic representation of microconversion events and the location of common mutations within the CYP21A2 gene transferred by gene conversion. NC, nonclassical; SV, simple virilizing; SW, salt-wasting.

Fig. 3. Gene rearrangement due to unequal crossover during meiosis. (A) CYP21A2 gene rearrangement caused by a 30-kb deletion, leading to a CYP21A1P/CYP21A2 chimera. The figure illustrates the 9 types of chimera according to the different junction sites. Seven chimeras harboring the I2G mutation are associated with the classic salt-wasting phenotype, and 2 chimeras carrying a p.P31L mutation exhibit an attenuated phenotype. (B) Formation of a chimeric TNXA/TNXB gene that deletes the CYP21A2 gene. Three different chimeric genes are demonstrated, depending on the junction site.

Fig. 4. Classification of common mutations in CYP21A2 based on their residual enzyme activity.
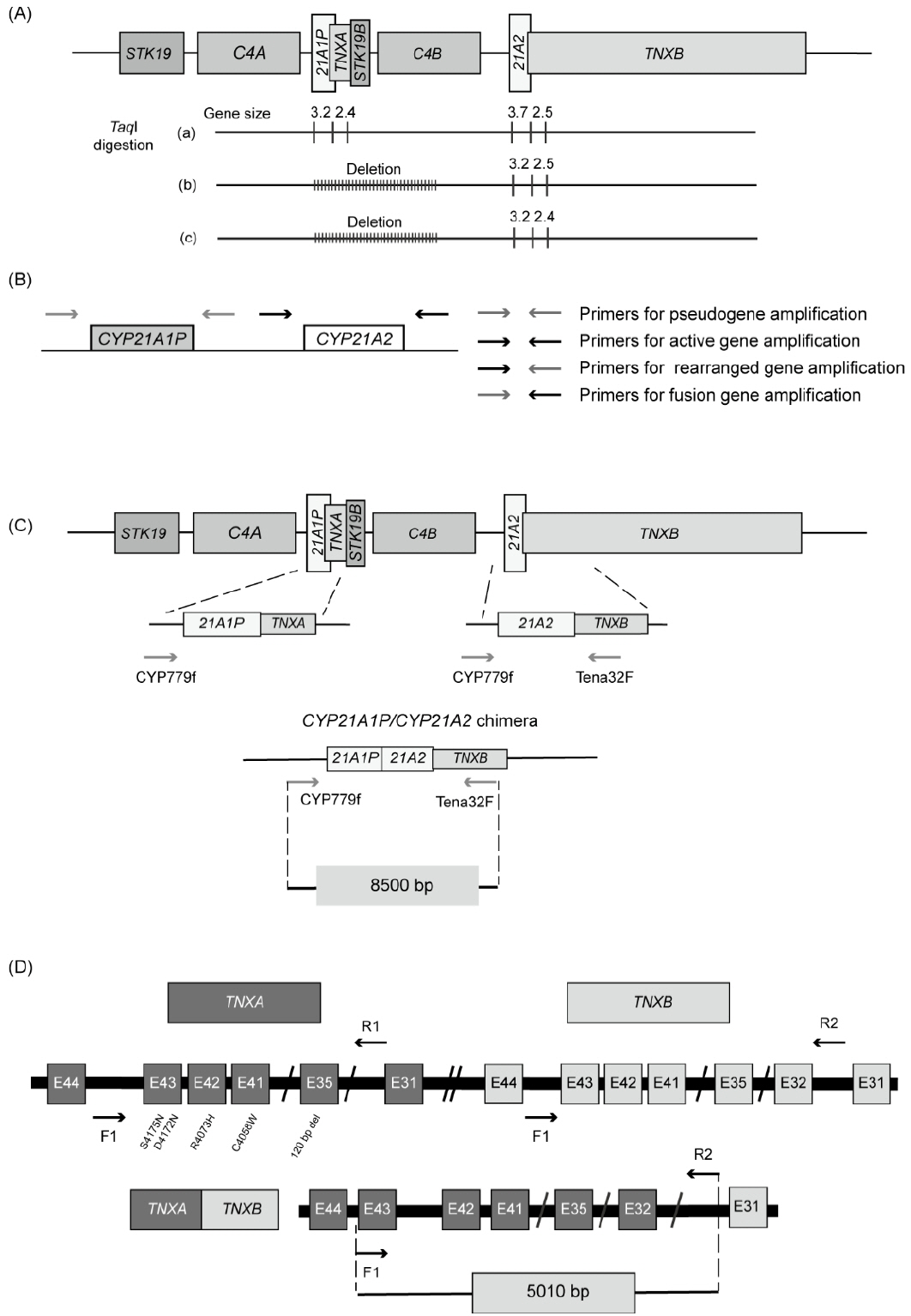
Fig. 5. Schematic representation of a molecular genetic analysis of CYP21A2. (A) Southern blot analysis digested with TaqI restriction endonuclease. This method distinguishes the CYP21A2 (3.7 kb) and CYP21A1P (3.2 kb) genes by fragment size. (B) Sequence-specific PCR amplification of the CYP21A1P and CYP21A2 genes. Two set of primers create 4 possible pairwise reactions to obtain amplicons corresponding to CYP21A1P, CYP21A2, the fusion gene, and rearrangement products.31) (C) Sequencing for a CYP21A1P/CYP21A2 chimera using paired primers, CYP779f/Tena32F. These primers amplify the 8.5-kb fragment. The Tena32F primer is located in a nonduplicated area of TNXB in exon 32, and the CYP779f primer is complementary to the 5’-end of the CYP21A1P and CYP21A2 genes. (D) Sequencing for a TNXA/TNXB chimera using mixed paired primers. The forward primer (F1) contains complementary sequences for intron 43 of TNXA and TNXB, and the reverse primer (R2) is specific for intron 31 of TNXB.
Reference
-
References
1. Claahsen-van der Grinten HL, Speiser PW, Ahmed SF, Arlt W, Auchus RJ, Falhammar H, et al. Congenital adrenal hyperplasia-Current insights in pathophysiology, diagnostics, and management. Endocr Rev. 2022; 43:91–159.2. Pignatelli D, Carvalho BL, Palmeiro A, Barros A, Guerreiro SG, Macut D. The complexities in genotyping of congenital adrenal hyperplasia: 21-hydroxylase deficiency. Front Endocrinol (Lausanne). 2019; 10:432.3. Higashi Y, Yoshioka H, Yamane M, Gotoh O, Fujii-Kuriyama Y. Complete nucleotide sequence of two steroid 21-hydroxylase genes tandemly arranged in human chromosome: a pseudogene and a genuine gene. Proc Natl Acad Sci U S A. 1986; 83:2841–5.4. White PC, New MI, Dupont B. Structure of human steroid 21-hydroxylase genes. Proc Natl Acad Sci U S A. 1986; 83:5111–5.5. den Dunnen JT, Dalgleish R, Maglott DR, Hart RK, Greenblatt MS, McGowan-Jordan J, et al. HGVS recommendations for the description of sequence variants: 2016 update. Hum Mutat. 2016; 37:564–9.6. Morel Y, Bristow J, Gitelman SE, Miller WL. Transcript encoded on the opposite strand of the human steroid 21-hydroxylase/complement component C4 gene locus. Proc Natl Acad Sci U S A. 1989; 86:6582–6.7. Lee H-H. Chimeric CYP21P/CYP21 and TNXA/TNXB genes in the RCCX module. Mol Genet Metab. 2005; 84:4–8.8. Yin C, Zhu B, Zhang T, Liu T, Chen S, Liu Y, et al. Pharmacological targeting of STK19 inhibits oncogenic NRAS-driven melanomagenesis. Cell. 2019; 176:1113–27.e16.9. Tassabehji M, Strachan T, Anderson M, Campbell RD, Collier S, Lako M. Identification of a novel family of human endogenous retroviruses and characterization of one family member, HERV-K(C4), located in the complement C4 gene cluster. Nucleic Acids Res. 1994; 22:5211–7.10. Blanchong CA, Zhou B, Rupert KL, Chung EK, Jones KN, Sotos JF, et al. Deficiencies of human complement component C4a and C4b and heterozygosity in length variants of RP-C4-CYP21-TNX (Rccx) modules in Caucasians: the load of Rccx genetic diversity on major histocompatibility complex–associated disease. J Exp Med. 2000; 191:2183–96.11. Grandi N, Cadeddu M, Pisano MP, Esposito F, Blomberg J, Tramontano E. Identification of a novel HERV-K(HML10): comprehensive characterization and comparative analysis in non-human primates provide insights about HML10 proviruses structure and diffusion. Mob DNA. 2017; 8:15.12. Carroll MC. Complement and humoral immunity. Vaccine. 2008; 26:I28–33.13. Wijesuriya SD, Zhang G, Dardis A, Miller WL. Transcriptional regulatory elements of the human gene for cytochrome P450c21 (steroid 21-hydroxylase) lie within intron 35 of the linked C4B gene. J Biol Chem. 1999; 274:38097–106.14. Miller WL, Merke DP. Tenascin-X, Congenital adrenal hHyperplasia, and the CAH-X syndrome. Horm Res Paediatr. 2018; 89:352–61.15. White PC, Grossberger D, Onufer BJ, Chaplin DD, New MI, Dupont B, et al. Two genes encoding steroid 21-hydroxylase are located near the genes encoding the fourth component of complement in man. Proc Natl Acad Sci U S A. 1985; 82:1089–93.16. Bristow J, Tee MK, Gitelman SE, Mellon SH, Miller WL. Tenascin-X: a novel extracellular matrix protein encoded by the human XB gene overlapping P450c21B. J Cell Biol. 1993; 122:265–78.17. Choi JH, Kim GH, Yo o HW. R ecent advances in biochemical and molecular analysis of congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Ann Pediatr Endocrinol Metab. 2016; 21:1–6.18. Concolino P, Costella A. Congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency: a comprehensive focus on 233 pathogenic variants of CYP21A2 Gene. Mol Diagn Ther. 2018; 22:261–80.19. Chen W, Xu Z, Sullivan A, Finkielstain GP, Van Ryzin C, Merke DP, et al. Junction site analysis of chimeric CYP21A1P/CYP21A2 genes in 21-hydroxylase deficiency. Clin Chem. 2012; 58:421–30.20. Billerbeck AE, Mendonca BB, Pinto EM, Madureira G, Arnhold IJ, Bachega TA. Three novel mutations in CYP21 gene in Brazilian patients with the classical form of 21-hydroxylase deficiency due to a founder effect. J Clin Endocrinol Metab. 2002; 87:4314–7.21. de Carvalho DF, Miranda MC, Gomes LG, Madureira G, Marcondes JAM, Billerbeck AEC, et al. Molecular CYP21A2 diagnosis in 480 Brazilian patients with congenital adrenal hyperplasia before newborn screening introduction. Eur J Endocrinol. 2016; 175:107–16.22. Loidi L, Quinteiro C, Parajes S, Barreiro J, Lestón DG, Cabezas-Agrícola JM, et al. High variability in CYP21A2 mutated alleles in Spanish 21-hydroxylase deficiency patients, six novel mutations and a founder effect. Clin Endocrinol (Oxf). 2006; 64:330–6.23. Speiser PW, Dupont J, Zhu D, Serrat J, Buegeleisen M, Tusie-Luna MT, et al. Disease expression and molecular genotype in congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Invest. 1992; 90:584–95.24. Riedl S, Röhl FW, Bonfig W, Brämswig J, Richter-Unruh A, Fricke-Otto S, et al. Genotype/phenotype correlations in 538 congenital adrenal hyperplasia patients from Germany and Austria: discordances in milder genotypes and in screened versus prescreening patients. Endocr Connect. 2019; 8:86–94.25. Kocova M, Anastasovska V, Falhammar H. Clinical outcomes and characteristics of P30L mutations in congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocrine. 2020; 69:262–77.26. Araujo RS, Billerbeck AE, Madureira G, Mendonca BB, Bachega TA. Substitutions in the CYP21A2 promoter explain the simple-virilizing form of 21-hydroxylase deficiency in patients harbouring a P30L mutation. Clin Endocrinol (Oxf). 2005; 62:132–6.27. New MI, Abraham M, Gonzalez B, Dumic M, Razzaghy-Azar M, Chitayat D, et al. Genotype–phenotype correlation in 1,507 families with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Proc Natl Acad Sci U S A. 2013; 110:2611–6.28. Speiser PW, Arlt W, Auchus RJ, Baskin LS, Conway GS, Merke DP, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2018; 103:4043–88.29. Romdhane L, Kefi R, Azaiez H, Halim NB, Dellagi K, Abdelhak S. Founder mutations in Tunisia: implications for diagnosis in North Africa and Middle East. Orphanet J Rare Dis. 2012; 7:52.30. Kharrat M, Tardy Vr, M’Rad R, Maazoul F, Jemaa LB, Refaï M, et al. Molecular genetic analysis of Tunisian patients with a classic form of 21-hydroxylase deficiency: identification of four novel mutations and high prevalence of Q318X mutation. J Clin Endocrinol Metab. 2004; 89:368–74.31. Choi JH, Jin HY, Lee BH, Ko JM, Lee JJ, Kim GH, et al. Clinical phenotype and mutation spectrum of the CYP21A2 gene in patients with steroid 21-hydroxylase deficiency. Exp Clin Endocrinol Diabetes. 2012; 120:23–7.32. Burch GH, Gong Y, Liu W, Dettman RW, Curry CJ, Smith L, et al. Tenascin–X deficiency is associated with Ehlers–Danlos syndrome. Nat Genet. 1997; 17:104–8.33. Mao JR, Taylor G, Dean WB, Wagner DR, Afzal V, Lotz JC, et al. Tenascin-X deficiency mimics Ehlers-Danlos syndrome in mice through alteration of collagen deposition. Nat Genet. 2002; 30:421–5.34. Morissette R, Chen W, Perritt AF, Dreiling JL, Arai AE, Sachdev V, et al. Broadening the spectrum of Ehlers Danlos syndrome in patients with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2015; 100:E1143–52.35. Merke DP, Chen W, Morissette R, Xu Z, Ryzin CV, Sachdev V, et al. Tenascin-X haploinsufficiency associated with Ehlers-Danlos syndrome in patients with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2013; 98:E379–87.36. Lao Q, Brookner B, Merke DP. High-throughput screening for CYP21A1P-TNXA/TNXB chimeric genes responsible for Ehlers-Danlos syndrome in patients with congenital adrenal hyperplasia. J Mol Diagn. 2019; 21:924–31.37. Gitelman SE, Bristow J, Miller WL. Mechanism and consequences of the duplication of the human C4/P450c21/gene X locus. Mol Cell Biol. 1992; 12:2124–34.38. Marino R, Garrido NP, Ramirez P, Notaristéfano G, Moresco A, Touzon MS, et al. Ehlers-Danlos syndrome: Molecular and clinical characterization of TNXA/TNXB chimeras in congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2021; 106:e2789. –802.39. Carrozza C, Foca L, Paolis ED, Concolino P. Genes and pseudogenes: complexity of the RCCX locus and disease. Front Endocrinol (Lausanne). 2021; 12:709758.40. Lao Q, Mallappa A, Rueda Faucz F, Joyal E, Veeraraghavan P, Chen W, et al. A TNXB splice donor site variant as a cause of hypermobility type Ehlers–Danlos syndrome in patients with congenital adrenal hyperplasia. Mol Genet Genomic Med. 2021; 9:e1556.41. Lee HH, Lee YJ, Chao MC. Comparing the Southern blot method and polymerase chain reaction product analysis for chimeric RCCX detection in CYP21A2 deficiency. Anal Biochem. 2010; 399:293–8.42. Olney RC, Mougey EB, Wang J, Shulman DI, Sylvester JE. Using real-time, quantitative PCR for rapid genotyping of the steroid 21-hydroxylase gene in a North Florida population. J Clin Endocrinol Metab. 2002; 87:735–41.43. Ravichandran L, Varghese D, R P, S AH, Korula S, Thomas N, et al. Allele-specific and multiplex PCR based tools for cost-effective and comprehensive genetic testing in Congenital Adrenal Hyperplasia. MethodsX. 2022; 9:101748.44. Baumgartner-Parzer S, Witsch-Baumgartner M, Hoeppner W. EMQN best practice guidelines for molecular genetic testing and reporting of 21-hydroxylase deficiency. Eur J Hum Genet. 2020; 28:1341–67.45. Krone N, Arlt W. Genetics of congenital adrenal hyperplasia. Best Pract Res Clin Endocrinol Metab. 2009; 23:181–92.46. Lee HH, Chang SF, Tsai FJ, Tsai LP, Lin CY. Mutation of IVS2-12A/C>G in combination with 707-714delGAGACTAC in the CYP21 gene is caused by deletion of the C4-CYP21 repeat module with steroid 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2003; 88:2726–9.47. Beighton P, De Paepe A, Steinmann B, Tsipouras P, Wenstrup RJ. Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK). Am J Med Genet. 1998; 77:31–7.48. De Coster W, Van Broeckhoven C. Newest methods for detecting structural variations. Trends Biotechnol. 2019; 37:973–82.49. Logsdon GA, Vollger MR, Eichler EE. Long-read human genome sequencing and its applications. Nat Rev Genet. 2020; 21:597–614.50. Liu Y, Chen M, Liu J, Mao A, Teng Y, Yan H, et al. Comprehensive analysis of congenital adrenal hyperplasia using long-read sequencing. Clin Chem. 2022; 68:927–39.51. Tantirukdham N, Sahakitrungruang T, Chaisiwamongkol R, Pongpanich M, Srichomthong C, Assawapitaksakul A, et al. Long-read amplicon sequencing of the CYP21A2 in 48 Thai patients with steroid 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2022; 107:1939–47.52. Marino R, Ramirez P, Galeano J, Perez Garrido N, Rocco C, Ciaccio M, et al. Steroid 21-hydroxylase gene mutational spectrum in 454 Argentinean patients: genotype–phenotype correlation in a large cohort of patients with congenital adrenal hyperplasia. Clin Endocrinol (Oxf ). 2011; 75:427–35.53. Finkielstain GP, Chen W, Mehta SP, Fujimura FK, Hanna RM, Van Ryzin C, et al. Comprehensive genetic analysis of 182 unrelated families with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2011; 96:E161–72.54. Krone N, Braun A, Roscher AA, Knorr D, Schwarz HP. Predicting phenotype in steroid 21-hydroxylase deficiency? Comprehensive genotyping in 155 unrelated, well defined patients from Southern Germany. J Clin Endocrinol Metab. 2000; 85:1059–65.55. Wang R, Yu Y, Ye J, Han L, Qiu W, Zhang H, et al. 21-hydroxylase deficiency-induced congenital adrenal hyperplasia in 230 Chinese patients: Genotype-phenotype correlation and identification of nine novel mutations. Steroids. 2016; 108:47–55.56. Koyama S, Toyoura T, Saisho S, Shimozawa K, Yata J. Genetic analysis of Japanese patients with 21-hydroxylase deficiency: identification of a patient with a new mutation of a homozygous deletion of adenine at codon 246 and patients without demonstrable mutations within the structural gene for CYP21. J Clin Endocrinol Metab. 2002; 87:2668–73.
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Clinical and molecular review of atypical congenital adrenal hyperplasia
- Recent advances in biochemical and molecular analysis of congenital adrenal hyperplasia due to 21-hydroxylase deficiency
- Management issues of congenital adrenal hyperplasia during the transition from pediatric to adult care
- Compound heterozygosity for a whole gene deletion and p.R124C mutation in CYP21A2 causing nonclassic congenital adrenal hyperplasia
- Duplication and deletion of 21 hydroxylase gene among the normal Korean subjects and in adrenogenital syndrome patients
