Alport syndrome: new advances in the last decade
- Kim JH
 1,2
1,2
- Affiliations
-
- 1Department of Pediatrics, Seoul National University Bundang Hospital, Seongnam, Republic of Korea
- 2Department of Pediatrics, Seoul National University College of Medicine, Seoul, Republic of Korea
- KMID: 2531932
- DOI: http://doi.org/10.3339/ckd.22.022
Abstract
- Alport syndrome (AS) is a progressive hereditary nephritis that is often accompanied by sensorineural hearing loss and ocular abnormalities. It is inherited in three modes of X-linked (XLAS), autosomal recessive (ARAS), and autosomal dominant (ADAS). XLAS is caused by pathogenic variants in COL4A5, while ARAS and ADAS are caused by those in COL4A3 or COL4A4. There is currently no curative treatment for AS; however, angiotensin-converting enzyme inhibitors (ACEi) can improve the outcome of AS. In the past decade, multiple studies have shown that early intervention with ACEi upon isolated microscopic hematuria or microalbuminuria could delay disease progression, and early diagnosis is crucial for early treatment. Therefore, a new classification of AS based on molecular diagnoses has been proposed, including the paradigm shift of re-classifying female “carriers” to “patients” and “thin basement membrane nephropathy” to “ADAS.” In addition, with the detection of COL4A mutations in some patients with biopsy-confirmed IgA nephropathy, focal segmental glomerulosclerosis, and chronic kidney disease of unknown origin, it is suggested that the phenotype of AS should be expanded. In this review, we highlight the landmark studies and guidelines published over the past decade and introduce strategies for early diagnosis and treatment to improve the outcomes of AS.
Keyword
Figure
-
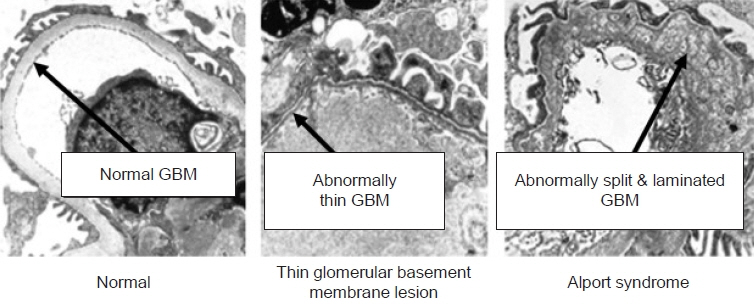
Fig. 1. Electron microscopy findings: the glomerular basement membrane (GBM) abnormalities caused by COL4A mutations. The GBM in Alport syndrome (AS) patients demonstrates irregular thickening with abnormal splitting and lamination. In patients with a thin basement membrane lesion, which is currently proposed to be regarded as AS, the GBM shows abnormally diffuse thinning. Reused from Warady et al. [20]. Images were used with permission from J. Charles Jennette. www.unckidneycenter.org. Accessed March 9, 2022.
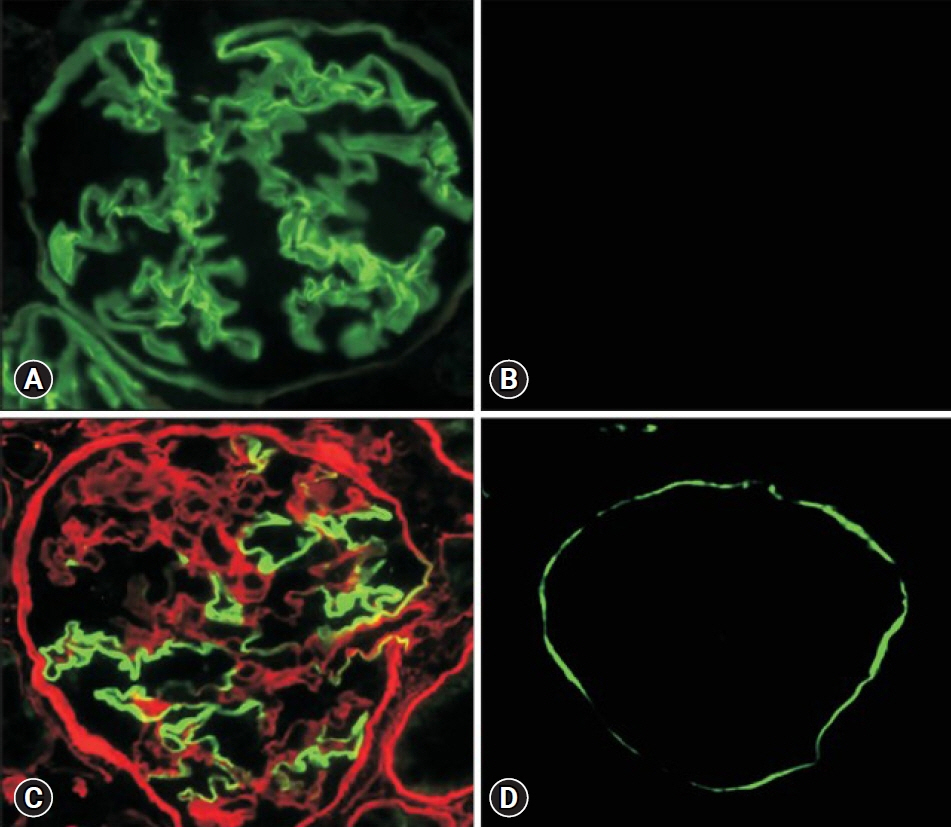
Fig. 2. Immunofluorescence staining of type IV collagen in glomerulus. (A) Normal control exhibits full expression in both the glomerular basement membrane (GBM) and Bowman capsule (BC). (B) An X-linked Alport syndrome (XLAS) male patient shows entirely negative expression in both GBM and BC. (C) An XLAS female patient displays a mosaic pattern of expression in both GBM and BC. Green and red colors indicate α5 and α2 chains of type IV collagen, respectively. (D) An autosomal recessive AS patient presents negative expression only on GBM, but positivity on BC. Reused from Nozu et al. [1].
Cited by 1 articles
-
Biomarkers for the early diagnosis of Alport syndrome and associated kidney damage
Hong Duc Thi Nguyen, Min Hyun Cho
Child Kidney Dis. 2025;29(1):12-18. doi: 10.3339/ckd.25.004.
Reference
-
References
1. Nozu K, Takaoka Y, Kai H, Takasato M, Yabuuchi K, Yamamura T, et al. Genetic background, recent advances in molecular biology, and development of novel therapy in Alport syndrome. Kidney Res Clin Pract. 2020; 39:402–13.
Article2. Kashtan CE, Gross O. Clinical practice recommendations for the diagnosis and management of Alport syndrome in children, adolescents, and young adults-an update for 2020. Pediatr Nephrol. 2021; 36:711–9.
Article3. Savige J, Ariani F, Mari F, Bruttini M, Renieri A, Gross O, et al. Expert consensus guidelines for the genetic diagnosis of Alport syndrome. Pediatr Nephrol. 2019; 34:1175–89.
Article4. Levey AS, Eckardt KU, Dorman NM, Christiansen SL, Hoorn EJ, Ingelfinger JR, et al. Nomenclature for kidney function and disease: report of a Kidney Disease: Improving Global Outcomes (KDIGO) consensus conference. Kidney Int. 2020; 97:1117–29.5. Nozu K, Nakanishi K, Abe Y, Udagawa T, Okada S, Okamoto T, et al. A review of clinical characteristics and genetic backgrounds in Alport syndrome. Clin Exp Nephrol. 2019; 23:158–68.
Article6. Boutaud A, Borza DB, Bondar O, Gunwar S, Netzer KO, Singh N, et al. Type IV collagen of the glomerular basement membrane: evidence that the chain specificity of network assembly is encoded by the noncollagenous NC1 domains. J Biol Chem. 2000; 275:30716–24.7. Bekheirnia MR, Reed B, Gregory MC, McFann K, Shamshirsaz AA, Masoumi A, et al. Genotype-phenotype correlation in X-linked Alport syndrome. J Am Soc Nephrol. 2010; 21:876–83.
Article8. Jais JP, Knebelmann B, Giatras I, Marchi M, Rizzoni G, Renieri A, et al. X-linked Alport syndrome: natural history in 195 families and genotype- phenotype correlations in males. J Am Soc Nephrol. 2000; 11:649–57.9. Gross O, Netzer KO, Lambrecht R, Seibold S, Weber M. Meta-analysis of genotype-phenotype correlation in X-linked Alport syndrome: impact on clinical counselling. Nephrol Dial Transplant. 2002; 17:1218–27.
Article10. Jais JP, Knebelmann B, Giatras I, De Marchi M, Rizzoni G, Renieri A, et al. X-linked Alport syndrome: natural history and genotype-phenotype correlations in girls and women belonging to 195 families: a "European Community Alport Syndrome Concerted Action" study. J Am Soc Nephrol. 2003; 14:2603–10.
Article11. Yamamura T, Nozu K, Fu XJ, Nozu Y, Ye MJ, Shono A, et al. Natural history and genotype-phenotype correlation in female X-linked Alport syndrome. Kidney Int Rep. 2017; 2:850–5.
Article12. Lee JM, Nozu K, Choi DE, Kang HG, Ha IS, Cheong HI. Features of autosomal recessive Alport syndrome: a systematic review. J Clin Med. 2019; 8:178.
Article13. Oka M, Nozu K, Kaito H, Fu XJ, Nakanishi K, Hashimura Y, et al. Natural history of genetically proven autosomal recessive Alport syndrome. Pediatr Nephrol. 2014; 29:1535–44.
Article14. Kamiyoshi N, Nozu K, Fu XJ, Morisada N, Nozu Y, Ye MJ, et al. Genetic, clinical, and pathologic backgrounds of patients with autosomal dominant Alport syndrome. Clin J Am Soc Nephrol. 2016; 11:1441–9.
Article15. Kashtan CE. Alport syndrome: an inherited disorder of renal, ocular, and cochlear basement membranes. Medicine (Baltimore). 1999; 78:338–60.
Article16. Sessa A, Meroni M. Alport's syndrome: Orphanet encyclopedia [Internet]. Paris: Orphanet; 2001 [cited 2022 Mar 2]. Available from: https://www.orpha.net.17. Savige J, Sheth S, Leys A, Nicholson A, Mack HG, Colville D. Ocular features in Alport syndrome: pathogenesis and clinical significance. Clin J Am Soc Nephrol. 2015; 10:703–9.
Article18. Adam J, Connor TM, Wood K, Lewis D, Naik R, Gale DP, et al. Genetic testing can resolve diagnostic confusion in Alport syndrome. Clin Kidney J. 2014; 7:197–200.
Article19. Yao XD, Chen X, Huang GY, Yu YT, Xu ST, Hu YL, et al. Challenge in pathologic diagnosis of Alport syndrome: evidence from correction of previous misdiagnosis. Orphanet J Rare Dis. 2012; 7:100.
Article20. Warady BA, Agarwal R, Bangalore S, Chapman A, Levin A, Stenvinkel P, et al. Alport syndrome classification and management. Kidney Med. 2020; 2:639–49.
Article21. Heidet L, Gubler MC. The renal lesions of Alport syndrome. J Am Soc Nephrol. 2009; 20:1210–5.
Article22. Hashimura Y, Nozu K, Kaito H, Nakanishi K, Fu XJ, Ohtsubo H, et al. Milder clinical aspects of X-linked Alport syndrome in men positive for the collagen IV α5 chain. Kidney Int. 2014; 85:1208–13.
Article23. Nakanishi K, Yoshikawa N, Iijima K, Kitagawa K, Nakamura H, Ito H, et al. Immunohistochemical study of alpha 1-5 chains of type IV collagen in hereditary nephritis. Kidney Int. 1994; 46:1413–21.24. IgA nephropathy combined with thin basement membrane nephropathy in children. Kidney Res Clin Pract. 2013; 32:194–5.25. Lennon R, Fornoni A. Could this be Alport syndrome? Clin J Am Soc Nephrol. 2021; 16:1743–5.
Article26. Savige J, Storey H, Watson E, Hertz JM, Deltas C, Renieri A, et al. Consensus statement on standards and guidelines for the molecular diagnostics of Alport syndrome: refining the ACMG criteria. Eur J Hum Genet. 2021; 29:1186–97.
Article27. Yamamura T, Horinouchi T, Nagano C, Omori T, Sakakibara N, Aoto Y, et al. Genotype-phenotype correlations influence the response to angiotensin-targeting drugs in Japanese patients with male X-linked Alport syndrome. Kidney Int. 2020; 98:1605–14.
Article28. Gross O, Beirowski B, Koepke ML, Kuck J, Reiner M, Addicks K, et al. Preemptive ramipril therapy delays renal failure and reduces renal fibrosis in COL4A3-knockout mice with Alport syndrome. Kidney Int. 2003; 63:438–46.29. Gross O, Licht C, Anders HJ, Hoppe B, Beck B, Tonshoff B, et al. Early angiotensin-converting enzyme inhibition in Alport syndrome delays renal failure and improves life expectancy. Kidney Int. 2012; 81:494–501.
Article30. Gross O, Tonshoff B, Weber LT, Pape L, Latta K, Fehrenbach H, et al. A multicenter, randomized, placebo-controlled, double-blind phase 3 trial with open-arm comparison indicates safety and efficacy of nephroprotective therapy with ramipril in children with Alport's syndrome. Kidney Int. 2020; 97:1275–86.
Article31. Groopman EE, Marasa M, Cameron-Christie S, Petrovski S, Aggarwal VS, Milo-Rasouly H, et al. Diagnostic utility of exome sequencing for kidney disease. N Engl J Med. 2019; 380:142–51.
Article32. Gast C, Pengelly RJ, Lyon M, Bunyan DJ, Seaby EG, Graham N, et al. Collagen (COL4A) mutations are the most frequent mutations underlying adult focal segmental glomerulosclerosis. Nephrol Dial Transplant. 2016; 31:961–70.33. Kashtan CE, Ding J, Garosi G, Heidet L, Massella L, Nakanishi K, et al. Alport syndrome: a unified classification of genetic disorders of collagen IV α345. A position paper of the Alport Syndrome Classification Working Group. Kidney Int. 2018; 93:1045–51.
Article34. Grunfeld JP, Noel LH, Hafez S, Droz D. Renal prognosis in women with hereditary nephritis. Clin Nephrol. 1985; 23:267–71.35. Rana K, Wang YY, Powell H, Jones C, McCredie D, Buzza M, et al. Persistent familial hematuria in children and the locus for thin basement membrane nephropathy. Pediatr Nephrol. 2005; 20:1729–37.
Article36. Imafuku A, Nozu K, Sawa N, Nakanishi K, Ubara Y. How to resolve confusion in the clinical setting for the diagnosis of heterozygous COL4A3 or COL4A4 gene variants? Discussion and suggestions from nephrologists. Clin Exp Nephrol. 2020; 24:651–6.
Article37. Yamamura T, Nozu K, Minamikawa S, Horinouchi T, Sakakibara N, Nagano C, et al. Comparison between conventional and comprehensive sequencing approaches for genetic diagnosis of Alport syndrome. Mol Genet Genomic Med. 2019; 7:e883.
Article38. Kashtan CE, Ding J, Gregory M, Gross O, Heidet L, Knebelmann B, et al. Clinical practice recommendations for the treatment of Alport syndrome: a statement of the Alport Syndrome Research Collaborative. Pediatr Nephrol. 2013; 28:5–11.
Article39. ESCAPE Trial Group, Wuhl E, Trivelli A, Picca S, Litwin M, Peco-Antic A, et al. Strict blood-pressure control and progression of renal failure in children. N Engl J Med. 2009; 361:1639–50.
Article40. Nakanishi K, Iijima K, Ishikura K, Hataya H, Awazu M, Sako M, et al. Efficacy and safety of lisinopril for mild childhood IgA nephropathy: a pilot study. Pediatr Nephrol. 2009; 24:845–9.
Article41. Gipson DS, Trachtman H, Kaskel FJ, Greene TH, Radeva MK, Gassman JJ, et al. Clinical trial of focal segmental glomerulosclerosis in children and young adults. Kidney Int. 2011; 80:868–78.
Article42. Gross O, Schulze-Lohoff E, Koepke ML, Beirowski B, Addicks K, Bloch W, et al. Antifibrotic, nephroprotective potential of ACE inhibitor vs AT1 antagonist in a murine model of renal fibrosis. Nephrol Dial Transplant. 2004; 19:1716–23.
Article43. Webb NJ, Shahinfar S, Wells TG, Massaad R, Gleim GW, McCrary Sisk C, et al. Losartan and enalapril are comparable in reducing proteinuria in children with Alport syndrome. Pediatr Nephrol. 2013; 28:737–43.
Article44. van den Belt SM, Heerspink HJL, Gracchi V, de Zeeuw D, Wuhl E, Schaefer F, et al. Early proteinuria lowering by angiotensin-converting enzyme inhibition predicts renal survival in children with CKD. J Am Soc Nephrol. 2018; 29:2225–33.
Article45. Kaito H, Nozu K, Iijima K, Nakanishi K, Yoshiya K, Kanda K, et al. The effect of aldosterone blockade in patients with Alport syndrome. Pediatr Nephrol. 2006; 21:1824–9.
Article46. Faul C, Donnelly M, Merscher-Gomez S, Chang YH, Franz S, Delfgaauw J, et al. The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat Med. 2008; 14:931–8.
Article47. Naesens M, Kuypers DR, Sarwal M. Calcineurin inhibitor nephrotoxicity. Clin J Am Soc Nephrol. 2009; 4:481–508.
Article48. Callis L, Vila A, Carrera M, Nieto J. Long-term effects of cyclosporine A in Alport's syndrome. Kidney Int. 1999; 55:1051–6.
Article49. Callis L, Vila A, Nieto J, Fortuny G. Effect of cyclosporin A on proteinuria in patients with Alport's syndrome. Pediatr Nephrol. 1992; 6:140–4.
Article50. Charbit M, Gubler MC, Dechaux M, Gagnadoux MF, Grunfeld JP, Niaudet P. Cyclosporin therapy in patients with Alport syndrome. Pediatr Nephrol. 2007; 22:57–63.
Article51. Massella L, Muda AO, Legato A, Di Zazzo G, Giannakakis K, Emma F. Cyclosporine A treatment in patients with Alport syndrome: a single-center experience. Pediatr Nephrol. 2010; 25:1269–75.
Article52. Kim JH, Lee DH, Lee B, Lim SH, Ahn YH, Kang HG, et al. Renal syndromic hearing loss is common in childhood-onset chronic kidney disease. J Korean Med Sci. 2020; 35:e364.
Article53. Alves FR, de A Quintanilha Ribeiro F. Revision about hearing loss in the Alport's syndrome, analyzing the clinical, genetic and bio-molecular aspects. Braz J Otorhinolaryngol. 2005; 71:813–9.
Article54. Zhang X, Zhang Y, Zhang Y, Gu H, Chen Z, Ren L, et al. X-linked Alport syndrome: pathogenic variant features and further auditory genotype-phenotype correlations in males. Orphanet J Rare Dis. 2018; 13:229.
Article55. Reata Pharmaceuticals. Reata announces positive results from year 2 of the pivotal phase 3 CARDINAL study of bardoxolone methyl in patients with Alport syndrome [Internet]. Reata Plano: Pharmaceuticals;2020. [cited 2022 Mar 5]. Available from: https://www.reatapharma.com/investors/news/news-details/2020/Reata-Announces-Positive-Results-From-Year-2-of-the-Pivotal-Phase-3-CARDINAL-Study-of-Bardoxolone-Methyl-in-Patients-with-Alport-Syndrome/default.aspx.56. de Zeeuw D, Akizawa T, Audhya P, Bakris GL, Chin M, Christ-Schmidt H, et al. Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N Engl J Med. 2013; 369:2492–503.
Article57. Gomez IG, MacKenna DA, Johnson BG, Kaimal V, Roach AM, Ren S, et al. Anti-microRNA-21 oligonucleotides prevent Alport nephropathy progression by stimulating metabolic pathways. J Clin Invest. 2015; 125:141–56.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- A Case of Anterior Lenticonus in Alport's Syndrome
- Diffuse Esophageal Leiomyomatosis in a Child with Alport Syndrome: Case Report
- Alport Syndrome Associated with Poststreptococcal Glomerulonephritis in Brothers
- Bilateral Serous Retinal Detachment Associated With Alport's Syndrome
- Two cases of Alport syndrome developed in sister
