Major Contribution of GREB1L Alterations to Severe Inner Ear Malformation Largely in a Non-mendelian Fashion
- Affiliations
-
- 1Department of Otolaryngology-Head and Neck Surgery, Chungnam National University Sejong Hospital, Chungnam National University College of Medicine, Sejong, Korea
- 2Department of Otorhinolaryngology-Head and Neck Surgery, Seoul National University Bundang Hospital, Seoul National University College of Medicine, Seongnam, Korea
- 3Department of Otorhinolaryngology-Head and Neck Surgery, Seoul National University Hospital, Seoul National University College of Medicine, Seoul, Korea
- 43billion Inc., Seoul, Korea
- KMID: 2527120
- DOI: http://doi.org/10.21053/ceo.2021.01935
Figure
-
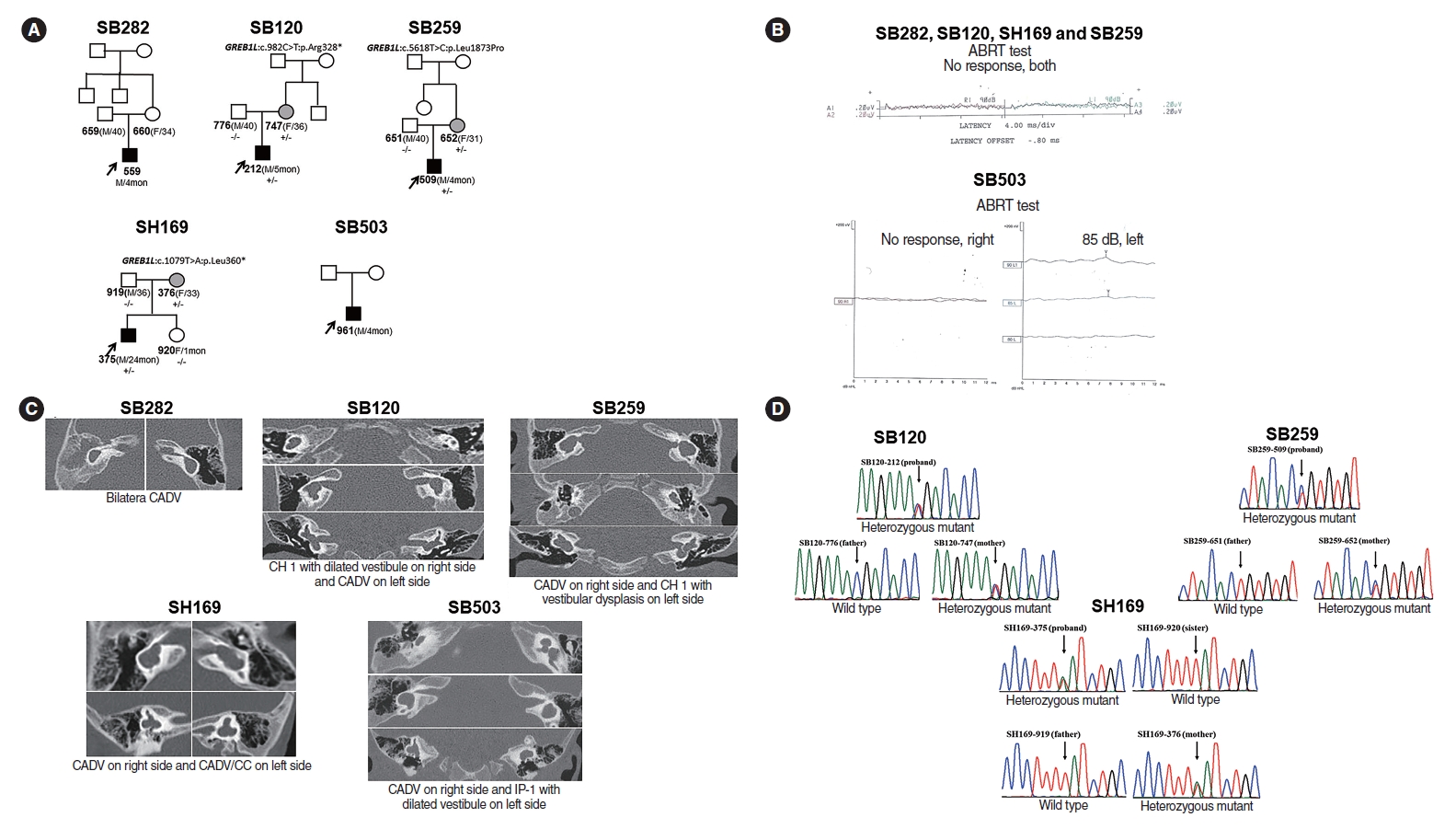
Fig. 1. Pedigrees, genotypes, and phenotypes of the five probands. (A) Black-filled symbols represent hearing-impaired individuals, clear symbols denote individuals with normal hearing, and gray-filled symbols indicate unaffected individuals who are heterozygous for the causative GREB1L variant in the pedigree (non-Mendelian inheritance). Black arrows represent the probands. (B) Auditory brain stem response threshold (ABRT) testing showed no response on both sides in all individuals except SB503, with 85 dB on the left side. (C) Temporal bone computed tomography revealed bilateral inner ear malformations. (D) A Sanger sequencing chromatogram confirmed the presence of each potential causative variant of GREB1L in the SB120, SB259, and SH169 pedigrees. CADV, cochlear aplasia with dilated vestibule; CH 1, cochlear hypoplasia type 1; CC, common cavity; IP-1, incomplete partition type 1.
Cited by 1 articles
-
How to Maximize the Outcomes of Cochlear Implantation in Common Cavity and Cochlear Aplasia With Dilated Vestibule, the Most Severe Inner Ear Anomalies?
Bong Jik Kim, Byung Yoon Choi
Clin Exp Otorhinolaryngol. 2022;15(1):3-4. doi: 10.21053/ceo.2022.00164.
Reference
-
1. Sennaroğlu L, Bajin MD. Classification and current management of inner ear malformations. Balkan Med J. 2017; Sep. 34(5):397–411.
Article2. Schrauwen I, Kari E, Mattox J, Llaci L, Smeeton J, Naymik M, et al. De novo variants in GREB1L areassociated with non-syndromic inner ear malformations and deafness. Hum Genet. 2018; Jul. 137(6-7):459–70.
Article3. Schrauwen I, Liaqat K, Schatteman I, Bharadwaj T, Nasir A, Acharya A, et al. Autosomal dominantly inherited GREB1L variants in individuals with profound sensorineural hearing impairment. Genes (Basel). 2020; Jun. 11(6):687.
Article4. De Tomasi L, David P, Humbert C, Silbermann F, Arrondel C, Tores F, et al. Mutations in GREB1L cause bilateral kidney agenesis in humans and mice. Am J Hum Genet. 2017; Nov. 101(5):803–14.
Article5. Kim BJ, Oh DY, Han JH, Oh J, Kim MY, Park HR, et al. Significant Mendelian genetic contribution to pediatric mild-to-moderate hearing loss and its comprehensive diagnostic approach. Genet Med. 2020; Jun. 22(6):1119–28.
Article6. Kim BJ, Kim AR, Lee C, Kim SY, Kim NK, Chang MY, et al. Discovery of CDH23 as a significant contributor to progressive postlingual sensorineural hearing loss in Koreans. PLoS One. 2016; Oct. 11(10):e0165680.
Article7. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015; May. 17(5):405–24.
Article8. Oza AM, DiStefano MT, Hemphill SE, Cushman BJ, Grant AR, Siegert RK, et al. Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum Mutat. 2018; Nov. 39(11):1593–613.
Article9. Kyei Barffour I, Kyei Baah Kwarkoh R. GREB1L as a candidate gene of Mayer-Rokitansky-Küster-Hauser Syndrome. Eur J Med Genet. 2021; Mar. 64(3):104158.
Article10. Rentzsch P, Schubach M, Shendure J, Kircher M. CADD-Splice-improving genome-wide variant effect prediction using deep learning-derived splice scores. Genome Med. 2021; Feb. 13(1):31.
Article11. Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet. 2016; Oct. 99(4):877–85.
Article12. Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020; May. 581(7809):434–43.
Article13. Jung KS, Hong KW, Jo HY, Choi J, Ban HJ, Cho SB, et al. KRGDB: the large-scale variant database of 1722 Koreans based on whole genome sequencing. Database (Oxford). 2020; Jan. 2020:baaa030.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Treatment of Secondary Macrotia Caused by Vascular Malformation: Report of Two Cases
- Exome Sequencing in Mendelian Disorders
- Accessory Earlobe Malformation
- Embolization of Dural Arteriovenous Malformation of the Jugular Bulb
- A Case of Multiple Pulmonary Arteriovenous Malformation Treated with Coil Embolization
