Balanced assessment of growth disorders using clinical, endocrinological, and genetic approaches
- Affiliations
-
- 1Centre for Endocrinology, William Harvey Research Institute, Barts and the London School of Medicine & Dentistry, Queen Mary, University of London, London, UK
- KMID: 2523827
- DOI: http://doi.org/10.6065/apem.2142208.104
Abstract
- Determining the pathogenesis of pediatric growth disorders is often challenging. In many cases, no pathogenesis is identified, and a designation of idiopathic short stature is used. The investigation of short stature requires a combination of clinical, endocrinological, and genetic evaluation. The techniques used are described, with equal importance being given to each of the 3 approaches. Clinical skills are essential to elicit an accurate history, family pedigree, and symptoms of body system dysfunction. Endocrine assessment requires hormonal determination for the diagnosis of hormone deficiency and initiation of successful replacement therapy. Genetic analysis has added a new dimension to the investigation of short stature and now uses next-generation sequencing with a candidate gene approach to confirm probable recognizable monogenic disorders and exome sequencing for complex phenotypes of unknown origin. Using the 3 approaches of clinical, endocrine, and genetic probes with equal status in the hierarchy of investigational variables provides the clinician with the highest chance of identifying the correct causative pathogenetic mechanism in a child presenting with short stature of unknown origin.
Keyword
Figure
-

Fig. 1. Scheme for clinical assessment of short stature.
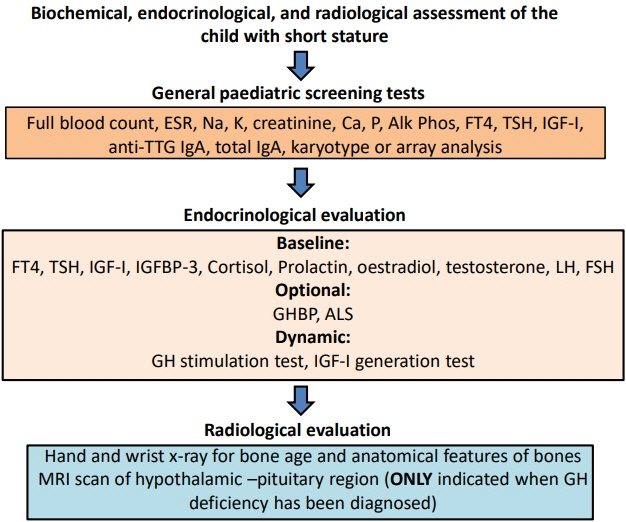
Fig. 2. Biochemical, endocrinological, and radiological assessment of the child with short stature. ESR, erythrocyte sedimentation rate; Alk Phos, alkaline phosphatase; FT4, Free thyroxine; TSH, thyroid-stimulating hormone; IGF-1, insulin-like growth factor-I; TTG, tissue transglutaminase; IGFBP-3, insulin-like growth factor binding protein-3; GHBP, growth hormone binding protein; ALS, acid labile subunit; MRI, magnetic resonance imaging; GH, growth hormone.
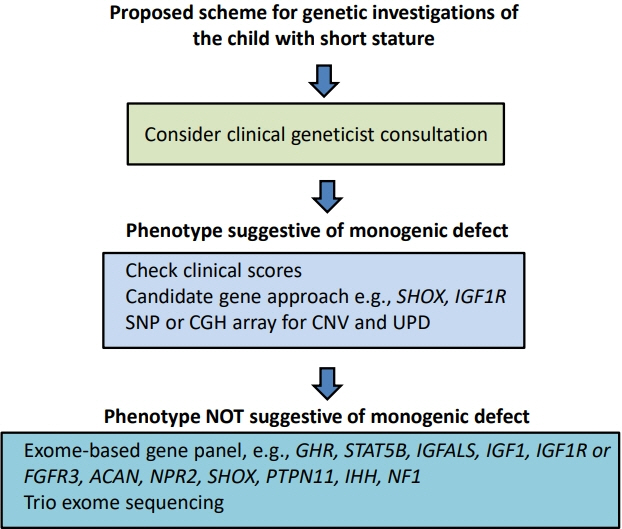
Fig. 3. Proposed scheme for genetic investigations of the child with short stature. SNP, single nucleotide polymorphism; CGH, comparative genomic hybridization; CNV, copy number variation; UPD, uniparental disomy.
Cited by 1 articles
-
Commentary on "Genetic evaluation using next-generation sequencing of children with short stature: a single tertiary-center experience"
Hye Young Jin
Ann Pediatr Endocrinol Metab. 2024;29(1):1-2. doi: 10.6065/apem.2423018edi01.
Reference
-
References
1. Hossain M, Choudhury N, Adib Binte Abdullah K, Mondal P, Jackson AA, Walson J, et al. Evidence-based approaches to childhood stunting in low and middle income countries: a systematic review. Arch Dis Child. 2017; 102:903–9.
Article2. Noeker M. Psychological functioning in idiopathic short stature. Horm Res Pediatr. 2011; 76 Suppl 3:52–6.
Article3. Savage MO, Backeljauw PF, Calzada R, Cianfarani S, Dunkel L, Koledova E, et al. Early detection, referral, investigation, and diagnosis of children with growth disorders. Horm Res Pediatr. 2016; 85:325–32.
Article4. Rapaport R, Wit JM, Savage MO. Growth failure: ‘idiopathic’ only after detailed investigation of short stature. ’ Endocr Connect. 2021; 10:R125–38.5. Baron J, Sävendahl L, De Luca F, Dauber A, Phillip M, Wit JM, et al. Short and tall stature: a new paradigm emerges. Nat Rev Endocrinol. 2015; 11:735–46.
Article6. Wit JM, Kamp GA, Oostdijk W; on behalf of the Dutch Working Group on Triage and Diagnosis of Growth Disorders in Children. Towards a rational and efficient diagnostic approach in children referred for growth failure to the general paediatrician. Horm Res Pediatr. 2019; 91:223–40.
Article7. Hermanussen M, Cole J. The calculation of target height reconsidered. Horm Res. 2003; 59:180–3.
Article8. Cohen P, Rogol AD, Deal CL, Saenger P, Reiter EO, Ross JL, et al. Consensus statement on the diagnosis and treatment of children with idiopathic short stature: a summary of the Growth Hormone Research Society, the Lawson Wilkins Pediatric Endocrine Society, and the European Society for Paediatric Endocrinology Workshop. J Clin Endocrinol Metab. 2008; 93:4210–7.
Article9. Sisley S, Trujillo MV, Khoury J, Backeljauw P. Low incidence of pathology detection and high cost of screening in the evaluation of asymptomatic short children. J Pediatr. 2013; 163:1045–51.
Article10. Dauber A. Genetic testing for the child with short stature-has the time come to change our diagnostic paradigm? J Clin Endocrinol Metab. 2019; 104:2766–9.
Article11. Wit JM, Clayton PE, Rogol AD, Savage MO, Saenger PH, Cohen P. Idiopathic short stature: definition, epidemiology, and diagnostic evaluation. Growth Horm IGF Res. 2008; 18:89–110.
Article12. Wit JM, Oostdijk W, Losekoot M, van Duyvenvoorde HA, Ruivenkamp CA, Kant SG. Mechanisms in endocrinology: novel genetic causes of short stature. Eur J Endocrinol. 2016; 174:R145–73.
Article13. Collett-Solberg PF, Ambler G, Backeljauw PF, Bidlingmaier M, Biller BMK, Boguszewski MCS, et al. Diagnosis, genetics, and therapy of short stature in children: a growth hormone research society international perspective. Horm Res Paediatr. 2019; 92:1–14.
Article14. Graber E, Rapaport R. Growth and growth disorders in children and adolescents. Pediatr Ann. 2012; 41:e1–9.
Article15. Blum WF, Alherbish A, Alsagheir A, El Awwa A, Kaplan W, Koledova E, et al. The growth hormone-insulin-like growth factor-I axis in the diagnosis and treatment of growth disorders. Endocr Connect. 2018; 7:R212–22.
Article16. Salmon WD, Daughaday WH. A hormonally controlled serum factor which stimulates sulfate incorporation by cartilage in vitro. J Lab Clin Med. 1957; 49:825–36.17. Kaplan SA, Cohen P. The somatomedin hypothesis 2007: 50 years later. J Clin Endocrinol Metab. 2007; 92:4529–35.18. LeRoith D. Clinical relevance of systemic and local IGF-I: lessons from animal models. Pediatr Endocrinol Rev. 2008; 5 Suppl 2:739–43.19. Ranke MB. Defining insulin-like growth factor-I deficiency. Horm Res. 2006; 65 Suppl 1:9–14.
Article20. Park P, Cohen P. Insulin-like growth factor (IGF-I) measurements in growth hormone (GH) therapy of idiopathic short stature (ISS). Growth Horm IGF Res. 2005; 15 Suppl A:S13–20.21. Cohen P, Rogol AD, Howard CP, Bright GM, Kappelgaard AM, Rosenfeld RG, et al. Insulin growth factor-based dosing of growth hormone therapy in children: a randomized, controlled study. J Clin Endocrinol Metab. 2007; 92:2480–6.
Article22. Selva KA, Buckway CK, Sexton G, Pratt KL, Tjoeng E, Guevara-Aguirre J, et al. Reproducibility in patterns of IGF generation with special reference to idiopathic short stature. Horm Res. 2003; 60:237–46.
Article23. Buckway CK, Guevara-Aguirre J, Pratt KL, Burren CP, Rosenfeld RG. The IGF-I generation test revisited: a marker of GH sensitivity. J Clin Endocrinol Metab. 2001; 86:5176–83.
Article24. Wit JM, Bidlingmaier M, de Bruin C, Oostdijk W. A Proposal for the Interpretation of S erum IGF-I concentration as part of laboratory screening in children with growth failure. J Clin Res Pediatr Endocrinol. 2020; 12:130–9.25. Yau M, Chacko E, Regelmann MO, Annunziato R, Wallach EJ, Chia D, et al. Peak growth hormone response to combined stimulation test in 315 children and correlations with metabolic parameters. Horm Res Paediatr. 2019; 92:36–44.
Article26. Coutant R, Dörr HG, Gleeson H, Argente J. Diagnosis of endocrine disease: limitations of the IGF1 generation test in children with short stature. Eur J Endocrinol. 2012; 166:351–7.
Article27. Argente J, Pérez-Jurado LA. Genetic causes of proportionate short stature. Best Pract Res Clin Endocrinol Metab. 2018; 32:499–522.
Article28. Sjoberg M, Salazar T, Espinosa C, Dagnino A, Avila A, Eggers M, et al. Study of GH sensitivity in Chilean patients with idiopathic short stature. J Clin Endocrinol Metab. 2001; 86:4375–81.
Article29. Storr HL, Chatterjee S, Metherell LA, Foley C, Rosenfeld RG, Backeljauw PF, et al. Nonclassical GH insensitivity: characterization of mild abnormalities of GH action. Endocr Rev. 2019; 40:476–505.
Article30. Vairamani K, Merjaneh L, Casano-Sancho P, Sanli ME, David A, Metherell LA, et al. Novel dominant-negative gh receptor mutations expands the spectrum of GHI and IGF-I deficiency. J Endocr Soc. 2017; 1:345–8.
Article31. Metherell LA, Akker SA, Munroe PB, Rose SJ, Caulfield M, Savage MO, et al. Pseudoexon activation as a novel mechanism for disease resulting in atypical growth-hormone insensitivity. Am J Hum Genet. 2001; 69:641–6.
Article32. Klammt J, Neumann D, Gevers EF, Andrew SF, Schwartz ID, Rockstroh D, et al. Dominant-negative STAT5B mutations cause growth hormone insensitivity with short stature and mild immune dysregulation. Nat Commun. 2018; 9:2105.
Article33. Heath KE, Argente J, Barrios V, Pozo J, Díaz-González F, Martos-Moreno GA, et al. Primary acid-labile subunit deficiency due to recessive IGFALS mutations results in postnatal growth deficit associated with low circulating insulin growth factor (IGF)-I, IGF binding protein-3 levels, and hyperinsulinemia. J Clin Endocrinol Metab. 2008; 93:1616–24.34. Dauber A, Muñoz-Calvo MT, Barrios V, Domené HM, Kloverpris S, Serra-Juhé C, et al. Mutations in pregnancy-associated plasma protein A2 cause short stature due to low IGF-I availability. EMBO Mol Med. 2016; 8:363–74.
Article35. Fofanova-Gambetti OV, Hwa V, Wit JM, Domene HM, Argente J, Bang P, et al. Impact of heterozygosity for acid-labile subunit (IGFALS) gene mutations on stature: results from the international acid-labile subunit consortium. J Clin Endocrinol Metab. 2010; 95:4184–91.
Article36. Walenkamp MJE, Robers JML, Wit JM, Zandwijken GRJ, van Duyvenvoorde HA, Oostdijk W, et al. Phenotypic features and response to GH treatment of patients with a molecular defect of the IGF-1 receptor. J Clin Endocrinol Metab. 2019; 104:3157–71.
Article37. Begemann M, Zirn B, Santen G, Wirthgen E, Soellner L, Büttel HM, et al. Paternally inherited IGF2 mutation and growth restriction. N Engl J Med. 2015; 373:349–56.
Article38. Finken MJJ, van der Steen M, Smeets CCJ, Walenkamp MJE, de Bruin C, Hokken-Koelega ACS, et al. Children born small for gestational age: differential diagnosis, molecular genetic evaluation, and implications. Endocr Rev. 2018; 39:851–94.
Article39. Fukami M, Seki A, Ogata T. SHOX haploinsufficiency as a cause of syndromic and nonsyndromic short stature. Mol Syndromol. 2016; 7:3–11.
Article40. Binder G. Short stature due to SHOX deficiency: genotype, phenotype, and therapy. Horm Res Paediatr. 2011; 75:81–9.
Article41. Benabbad I, Rosilio M, Child CJ, Carel JC, Ross JL, Deal CL, et al. Safety outcomes and near-adult height gain of growth hormone-treated children with shox deficiency: data from an observational study and a clinical trial. Horm Res Paediatr. 2017; 87:42–50.
Article42. Shapiro S, Klein GW, Klein ML, Wallach EJ, Fen Y, Godbold JH, et al. SHOX gene variants: growth hormone/insulin-like growth factor-1 status and response to growth hormone treatment. Horm Res Paediatr. 2015; 83:26–35.
Article43. Kant SG, Cervenkova I, Balek L, Trantirek L, Santen GW, de Vries MC, et al. A novel variant of FGFR3 causes proportionate short stature. Eur J Endocrinol. 2015; 172:763–70.
Article44. Pinto G, Cormier-Daire V, Le Merrer M, Samara-Boustani D, Baujat G, Fresneau L, et al. Efficacy and safety of growth hormone treatment in children with hypochondroplasia: comparison with an historical cohort. Horm Res Paediatr. 2014; 82:355–63.
Article45. Hisado-Oliva A, Garre-Vázquez AI, Santaolalla-Caballero F, Belinchón A, Barreda-Bonis AC, Vasques GA, et al. Heterozygous NPR2 mutations cause disproportionate short stature, similar to Léri-Weill dyschondrosteosis. J Clin Endocrinol Metab. 2015; 100:E1133–42.46. Vasques GA, Amano N, Docko AJ, Funari MF, Quedas EP, Nishi MY, et al. Heterozygous mutations in natriuretic peptide receptor-B (NPR2) gene as a cause of short stature in patients initially classified as idiopathic short stature. J Clin Endocrinol Metab. 2013; 98:E1636–44.47. Wang SR, Jacobsen CM, Carmichael H, Edmund AB, Robinson JW, Olney RC, et al. Heterozygous mutations in natriuretic peptide receptor-B (NPR2) gene as a cause of short stature. Hum Mutat. 2015; 36:474–81.48. Nilsson O, Guo MH, Dunbar N, Popovic J, Flynn D, Jacobsen C, et al. Short stature, accelerated bone maturation, and early growth cessation due to heterozygous aggrecan mutations. J Clin Endocrinol Metab. 2014; 99:E1510–8.
Article49. Hauer NN, Sticht H, B oppudi S, Büttner C, Kraus C, Trautmann U, et al. Genetic screening confirms heterozygous mutations in ACAN as a major cause of idiopathic short stature. Sci Rep. 2017; 7:12225.
Article50. Hauer NN, Popp B, Schoeller E, Schuhmann S, Heath KE, Hisado-Oliva A, et al. Clinical relevance of systematic phenotyping and exome sequencing in patients with short stature. Genet Med. 2018; 20:630–8.
Article51. Dauber A, Rosenfeld RG, Hirschhorn JN. Genetic evaluation of short stature. J Clin Endocrinol Metab. 2014; 99:3080–92.
Article52. Argente J, Tatton-Brown K, Lehwalder D, Pfäffle R. Genetics of growth disorders-which patients require genetic testing? Front Endocrinol (Lausanne). 2019; 10:602.
Article53. Murray PG, Clayton PE, Chernausek SD. A genetic approach to evaluation of short stature of undetermined cause. Lancet Diabetes Endocrinol. 2018; 6:564–74.
Article54. Wang SR, Carmichael H, Andrew SF, Miller TC, Moon JE, Derr MA, et al. Large-scale pooled next-generation sequencing of 1077 genes to identify genetic causes of short stature. J Clin Endocrinol Metab. 2013; 98:E1428–37.
Article55. Nilsson O. Aggrecanopathies highlight the need for genetic evaluation of ISS children. Eur J Endocrinol. 2020; 183:C9–10.
Article56. Chatterjee S, Shapiro L, Rose SJ, Mushtaq T, Clayton PE, Ten SB, et al. Phenotypic spectrum and responses to recombinant human IGF1 (rhIGF1) therapy in patients with homozygous intronic pseudoexon growth hormone receptor mutation. Eur J Endocrinol. 2018; 178:481–9.
Article57. Wakeling EL, Brioude F, Lokulo-Sodipe O, O'Connell SM, Salem J, Bliek J, et al. Diagnosis and management of SilverRussell syndrome: first international consensus statement. Nat Rev Endocrinol. 2017; 13:105–24.
Article58. Freire BL, Homma TK, Funari MFA, Lerario AM, Vasques GA, Malaquias AC, et al. Multigene sequencing analysis of children born small for gestational age with isolated short stature. J Clin Endocrinol Metab. 2019; 104:2023–30.
Article59. Cottrell E, Ladha T, Borysewicz-Sańczyk H, Sawicka B, Savage MO, Bossowski AT, et al. The value of whole exome sequencing for genetic diagnosis in a patient with Bloom syndrome. J Endocrinol Invest. 2021; 44:1331–4.
Article60. Stalman SE, Solanky N, Ishida M, Alemán-Charlet C, Abu-Amero S, Alders M, et al. Genetic analyses in small0for-gestational-age newborns. J Clin Endocrinol Metab. 2018; 103:917–25.61. Howell SJ, Wilton P, Lindberg A, Shalet SM. Growth hormone replacement and the risk of malignancy in children with neurofibromatosis. J Pediatr. 1998; 133:201–5.
Article62. Noseworthy J. The future of care - preserving the patient-physician relationship. N Engl J Med. 2019; 381:2265–9.
Article
