Yeungnam Univ J Med.
2021 Jul;38(3):208-218. 10.12701/yujm.2020.00591.
A retrospective analysis of etiology and outcomes of hemophagocytic lymphohistiocytosis in children and adults
- Affiliations
-
- 1Department of Pediatrics, Keimyung University Dongsan Hospital, Keimyung University School of Medicine, Daegu, Korea
- 2Department of Pediatrics, Keimyung University Daegu Dongsan Hospital, Daegu, Korea
- 3Department of Pediatrics, Yeungnam University Hospital, Daegu, Korea
- 4Department of Pediatrics, Yeungnam University College of Medicine, Daegu, Korea
- 5Department of Internal Medicine, Keimyung University Dongsan Hospital, Keimyung University School of Medicine, Daegu, Korea
- KMID: 2518662
- DOI: http://doi.org/10.12701/yujm.2020.00591
Abstract
- Background
Hemophagocytic lymphohistiocytosis (HLH) is a rare but severe, life-threatening inflammatory condition if untreated. We aimed to investigate the etiologies, outcomes, and risk factors for death in children and adults with HLH.
Methods
The medical records of patients who met the HLH criteria of two regional university hospitals in Korea between January 2001 and December 2019 were retrospectively investigated.
Results
Sixty patients with HLH (35 children and 25 adults) were included. The median age at diagnosis was 7.0 years (range, 0.1–83 years), and the median follow-up duration was 8.5 months (range, 0–204 months). Four patients had primary HLH, 48 patients had secondary HLH (20 infection-associated, 18 neoplasm-associated, and 10 autoimmune-associated HLH), and eight patients had HLH of unknown cause. Infection was the most common cause in children (14/35, 40.0%), whereas neoplasia was the most common cause in adults (13/25, 52.0%). Twenty-eight patients were treated with HLH-2004/94 immunochemotherapy. The 5-year overall survival (OS) rate for all HLH patients was 59.9%. The 5-year OS rates for patients with primary, infection-associated, neoplasm-associated, autoimmune-associated, and unknown cause HLH were 25.0%, 85.0%, 26.7%, 87.5%, and 62.5%, respectively. Using multivariate analysis, neoplasm-induced HLH (p=0.001) and a platelet count <50×109/L (p=0.008) were identified as independent risk factors for poor prognosis in patients with HLH.
Conclusion
Infection was the most common cause of HLH in children, while it was neoplasia in adults. The 5-year OS rate for all HLH patients was 59.9%. HLH caused by an underlying neoplasm or a low platelet count at the time of diagnosis were risk factors for poor prognosis.
Keyword
Figure
-
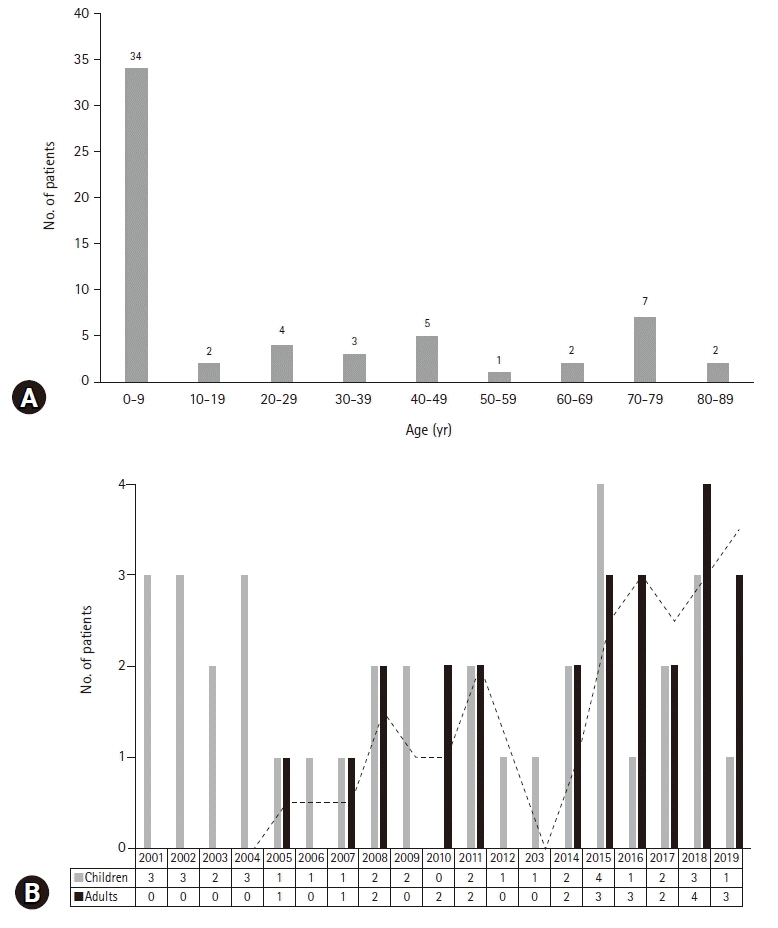
Fig. 1. (A) Age distribution of patients with hemophagocytic lymphohistiocytosis (HLH). (B) Number of patients diagnosed with HLH per year.
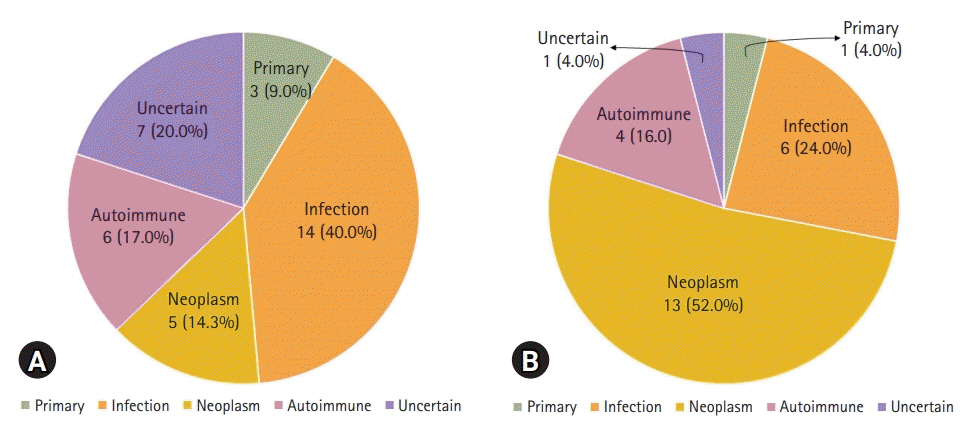
Fig. 2. Underlying causes of hemophagocytic lymphohistiocytosis according to the age groups. (A) Children, aged <18 years (n=35). (B) Adults, aged ≥18 years (n=25).

Fig. 3. (A) In all patients with hemophagocytic lymphohistiocytosis (HLH) (N=60), the 5-year overall survival (OS) rate is 59.9% (95% CI, 46.6–73.2). (B) The 5-year OS according to the classification of HLH.
Reference
-
References
1. Henter JI, Samuelsson-Horne A, Aricò M, Egeler RM, Elinder G, Filipovich AH, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. 2002; 100:2367–73.
Article2. Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis. Eur J Pediatr. 2007; 166:95–109.
Article3. Janka GE. Familial hemophagocytic lymphohistiocytosis. Eur J Pediatr. 1983; 140:221–30.
Article4. Henter JI, Aricò M, Elinder G, Imashuku S, Janka G. Familial hemophagocytic lymphohistiocytosis. Primary hemophagocytic lymphohistiocytosis. Hematol Oncol Clin North Am. 1998; 12:417–33.5. Jordan MB, Allen CE, Weitzman S, Filipovich AH, McClain KL. How I treat hemophagocytic lymphohistiocytosis. Blood. 2011; 118:4041–52.
Article6. Sieni E, Cetica V, Hackmann Y, Coniglio ML, Da Ros M, Ciambotti B, et al. Familial hemophagocytic lymphohistiocytosis: when rare diseases shed light on immune system functioning. Front Immunol. 2014; 5:167.
Article7. Aricò M, Janka G, Fischer A, Henter JI, Blanche S, Elinder G, et al. Hemophagocytic lymphohistiocytosis. Report of 122 children from the International Registry. FHL Study Group of the Histiocyte Society. Leukemia. 1996; 10:197–203.8. Henter JI, Elinder G, Ost A. Diagnostic guidelines for hemophagocytic lymphohistiocytosis. The FHL Study Group of the Histiocyte Society. Semin Oncol. 1991; 18:29–33.9. Henter JI, Aricò M, Egeler RM, Elinder G, Favara BE, Filipovich AH, et al. HLH-94: a treatment protocol for hemophagocytic lymphohistiocytosis. HLH study Group of the Histiocyte Society. Med Pediatr Oncol. 1997; 28:342–7.10. Trottestam H, Horne A, Aricò M, Egeler RM, Filipovich AH, Gadner H, et al. Chemoimmunotherapy for hemophagocytic lymphohistiocytosis: long-term results of the HLH-94 treatment protocol. Blood. 2011; 118:4577–84.
Article11. Henter JI, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007; 48:124–31.
Article12. Bergsten E, Horne A, Aricó M, Astigarraga I, Egeler RM, Filipovich AH, et al. Confirmed efficacy of etoposide and dexamethasone in HLH treatment: long-term results of the cooperative HLH-2004 study. Blood. 2017; 130:2728–38.
Article13. Koh KN, Im HJ, Chung NG, Cho B, Kang HJ, Shin HY, et al. Clinical features, genetics, and outcome of pediatric patients with hemophagocytic lymphohistiocytosis in Korea: report of a nationwide survey from Korea Histiocytosis Working Party. Eur J Haematol. 2015; 94:51–9.
Article14. Trizzino A, zur Stadt U, Ueda I, Risma K, Janka G, Ishii E, et al. Genotype-phenotype study of familial haemophagocytic lymphohistiocytosis due to perforin mutations. J Med Genet. 2008; 45:15–21.
Article15. Zur Stadt U, Beutel K, Kolberg S, Schneppenheim R, Kabisch H, Janka G, et al. Mutation spectrum in children with primary hemophagocytic lymphohistiocytosis: molecular and functional analyses of PRF1, UNC13D, STX11, and RAB27A. Hum Mutat. 2006; 27:62–8.16. Ramos-Casals M, Brito-Zerón P, López-Guillermo A, Khamashta MA, Bosch X. Adult haemophagocytic syndrome. Lancet. 2014; 383:1503–16.
Article17. Kleynberg RL, Schiller GJ. Secondary hemophagocytic lymphohistiocytosis in adults: an update on diagnosis and therapy. Clin Adv Hematol Oncol. 2012; 10:726–32.18. Lee H, Kim HS, Lee JM, Park KH, Choi AR, Yoon JH, et al. Natural killer cell function tests by flowcytometry-based cytotoxicity and IFN-γ production for the diagnosis of adult hemophagocytic lymphohistiocytosis. Int J Mol Sci. 2019; 20:5413.
Article19. Ishii E, Ohga S, Imashuku S, Yasukawa M, Tsuda H, Miura I, et al. Nationwide survey of hemophagocytic lymphohistiocytosis in Japan. Int J Hematol. 2007; 86:58–65.
Article20. La Rosée P, Horne A, Hines M, von Bahr Greenwood T, Machowicz R, Berliner N, et al. Recommendations for the management of hemophagocytic lymphohistiocytosis in adults. Blood. 2019; 133:2465–77.
Article21. Cetica V, Sieni E, Pende D, Danesino C, De Fusco C, Locatelli F, et al. Genetic predisposition to hemophagocytic lymphohistiocytosis: Report on 500 patients from the Italian registry. J Allergy Clin Immunol. 2016; 137:188–96.
Article22. Chen X, Wang F, Zhang Y, Teng W, Wang M, Nie D, et al. Genetic variant spectrum in 265 Chinese patients with hemophagocytic lymphohistiocytosis: molecular analyses of PRF1, UNC13D, STX11, STXBP2, SH2D1A, and XIAP. Clin Genet. 2018; 94:200–12.
Article23. Jin Z, Wang Y, Wang J, Zhang J, Wu L, Gao Z, et al. Primary hemophagocytic lymphohistiocytosis in adults: the utility of family surveys in a single-center study from China. Orphanet J Rare Dis. 2018; 13:17.
Article24. Hayden A, Park S, Giustini D, Lee AY, Chen LY. Hemophagocytic syndromes (HPSs) including hemophagocytic lymphohistiocytosis (HLH) in adults: a systematic scoping review. Blood Rev. 2016; 30:411–20.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- A Case of Hemophagocytic Lymphohistiocytosis Presenting with Neck Mass in a Child
- Multiple Ecthyma Gangrenosum in a Hemophagocytic Lymphohistiocytosis Patient
- Two Cases of Hemophagocytic Lymphohistiocytosis Following Kikuchi's Disease
- Hemophagocytic Lymphohistiocytosis
- Hemophagocytic lymphohistiocytosis secondary to histoplasmosis
