Ann Clin Neurophysiol.
2020 Oct;22(2):121-124. 10.14253/acn.2020.22.2.121.
Hereditary spastic paraplegia with thin corpus callosum due to novel homozygous mutation in SPG11 gene
- Affiliations
-
- 1Department of Neurology, Jeju National University School of Medicine, Jeju, Korea
- KMID: 2511089
- DOI: http://doi.org/10.14253/acn.2020.22.2.121
Abstract
- The most common form of autosomal recessive hereditary spastic paraplegia (HSP) is caused by mutations in SPG11/KIAA1840 gene, which encodes for spatacsin. The clinical presentation of SPG11 is characterized by cognitive impairment, peripheral neuropathy and a thin corpus callosum in brain magnetic resonance imaging. We identified a novel homozygous nonsense mutation (c.6082C>T [p.Q2028]) in exon 32 of SPG11 in Korean siblings. Our findings suggest that this novel homozygous mutation in SPG11 is associated with HSP and with dysgenesis of the corpus callosum.
Figure
-

Fig. 1. Magnetic resonance imaging findings for the proband. (A) Sagittal T1-weighted image showed thinning of the corpus callosum, which can be observed most prominently in the rostrum, genu, and body. (B) Axial T2-weighted image showed white matter changes surrounding the ventricle.
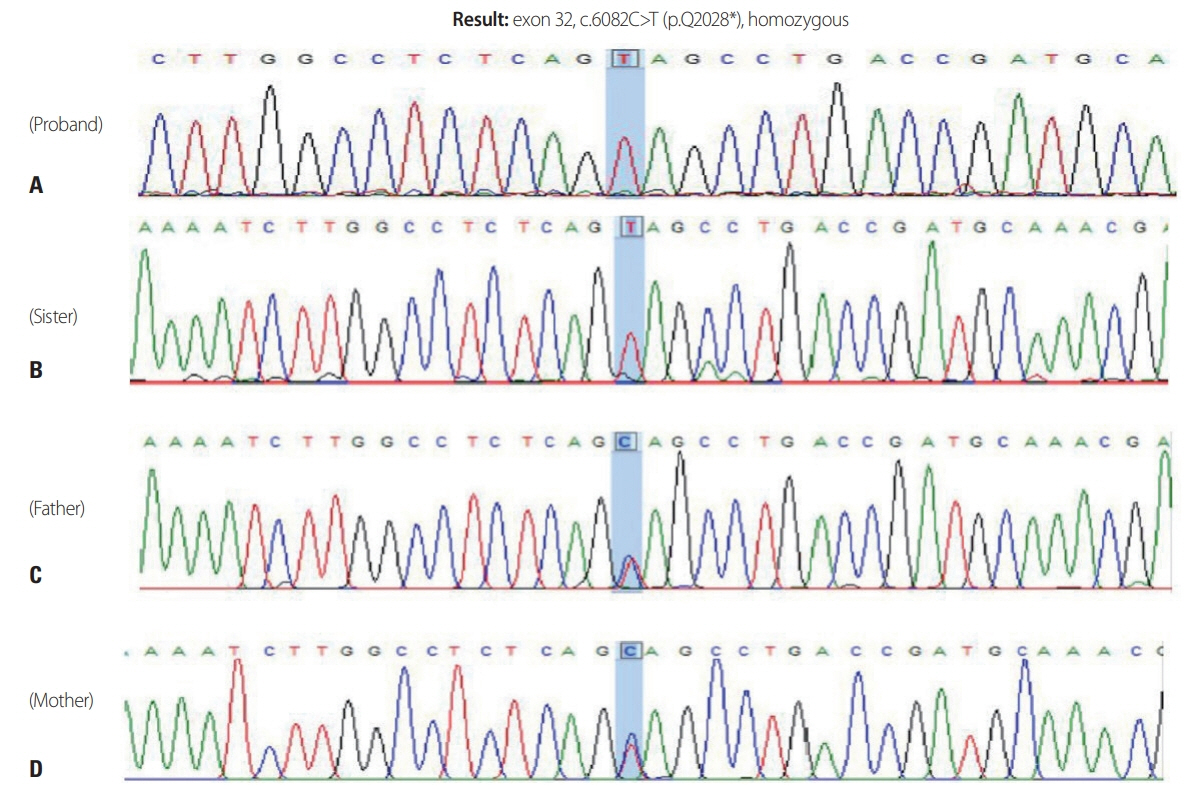
Fig. 2. Genetic analysis by polymerase chain reaction-Sanger sequencing of all exons of SPG11 and flanking regions of each exon revealed a homozygous nonsense mutation (c.6082C>T) in exon 32 (A). Sequencing analysis showed that both parents carried a heterozygous SPG11 c.6082C>T mutation (C, D), while the sister carried the same homozygous mutation as the proband (B).
Reference
-
1. Stevanin G, Santorelli FM, Azzedine H, Coutinho P, Chomilier J, Denora PS, et al. Mutations in SPG11, encoding spatacsin, are a major cause of spastic paraplegia with thin corpus callosum. Nat Genet. 2007; 39:366–372.
Article2. Stevanin G, Azzedine H, Denora P, Boukhris A, Tazir M, Lossos A, et al. Mutations in SPG11 are frequent in autosomal recessive spastic paraplegia with thin corpus callosum, cognitive decline and lower motor neuron degeneration. Brain. 2008; 131:772–784.
Article3. Finsterer J, Löscher W, Quasthoff S, Wanschitz J, Auer-Grumbach M, Stevanin G. Hereditary spastic paraplegia with autosomal dominant, recessive, X-linked, or maternal trait of inheritance. J Neurol Sci. 2012; 318:1–18.4. Salinas S, Proukakis C, Crosby A, Warner T. Hereditary spastic paraplegia: clinical features and pathogenetic mechanisms. Lancet Neurol. 2008; 7:1127–1138.
Article5. Shribman S, Reid E, Crosby AH, Houlden H, Warner TT. Hereditary spastic paraplegia: from diagnosis to emerging therapeutic approaches. Lancet Neurol. 2019; 18:1136–1146.
Article6. Kara E, Tucci A, Manzoni C, Lynch DS, Elpidorou M, Bettencourt C, et al. Genetic and phenotypic characterization of complex hereditary spastic paraplegia. Brain. 2016; 139(Pt 7):1904–1918.
Article7. Depienne C, Stevanin G, Brice A, Durr A. Hereditary spastic paraplegias: an update. Curr Opin Neurol. 2007; 20:674–680.
Article8. Hirst J, Borner GH, Edgar J, Hein MY, Mann M, Buchholz F, et al. Interaction between AP-5 and the hereditary spastic paraplegia proteins SPG11 and SPG15. Mol Biol Cell. 2013; 24:2558–2569.
Article9. Varga RE, Khundadze M, Damme M, Nietzsche S, Hoffmann B, Stauber T, et al. In vivo evidence for lysosome depletion and impaired autophagic clearance in hereditary spastic paraplegia type SPG11. PLoS Genet. 2015; 11:e1005454.
Article10. Branchu J, Boutry M, Sourd L, Depp M, Leone C, Corriger A, et al. Loss of spatacsin function alters lysosomal lipid clearance leading to upper and lower motor neuron degeneration. Neurobiol Dis. 2017; 102:21–37.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Thinning of the Corpus Callosum and Cerebellar Atrophy is Correlated with Phenotypic Severity in a Family with Spastic Paraplegia Type 11
- SPG11 Mutation in Hereditary Spastic Paraplegia with Thin Corpus Callosum Diagnosed by Targeted Gene Panel Sequencing
- Two novel mutations in ALDH18A1 and SPG11 gene found by whole-exome sequencing in spastic paraplegia disease patients in Iran
- Complicated Spastic Paraparesis with Thin Corpus Callosum
- Syringomyelia: A New Phenotype of SPG11-Related Hereditary Spastic Paraplegia?
