KCNQ2 Encephalopathy Showing a Distinct Ictal Amplitude-Integrated Electroencephalographic Pattern
- Affiliations
-
- 1Department of Pediatrics, School of Medicine, Kyungpook National University, Daegu, Korea
- KMID: 2510827
- DOI: http://doi.org/10.5385/nm.2020.27.4.202
Abstract
- KCNQ2 mutations induce a neonatal-onset epileptic encephalopathy of widely varying severity, ranging from benign familial neonatal epilepsy to severe refractory epileptic encephalopathy. Refractory seizures with KCNQ2 mutations have a positive response to sodium-channel blockers. Recently, a distinctive ictal pattern has been reported during amplitude-integrated electroencephalographic (aEEG) monitoring in infants with KCNQ2 encephalopathy. Herein, we describe a case of KCNQ2 encephalopathy with this distinctive ictal aEEG pattern, which was confirmed using conventional electroencephalography (EEG). A 3-day-old female infant presented with neonatal seizures accompanied by cyanosis and desaturation. Her seizure semiology was tonic and focal clonic. Her ictal aEEG demonstrated a sudden rise in amplitude followed by a suppressed background pattern. This pattern was also confirmed on conventional EEG. Her seizures were refractory despite the administration of multiple conventional antiepileptic drugs. Finally, c.794C>T; p. (Ala265Val) mutation was observed in the KCNQ2 gene on genetic testing, and she was diagnosed with KCNQ2 encephalopathy. Identifying this distinctive ictal pattern on aEEG monitoring facilitates the early detection of KCNQ2 encephalopathy and timely targeted treatment in patients with refractory seizures.
Figure
-
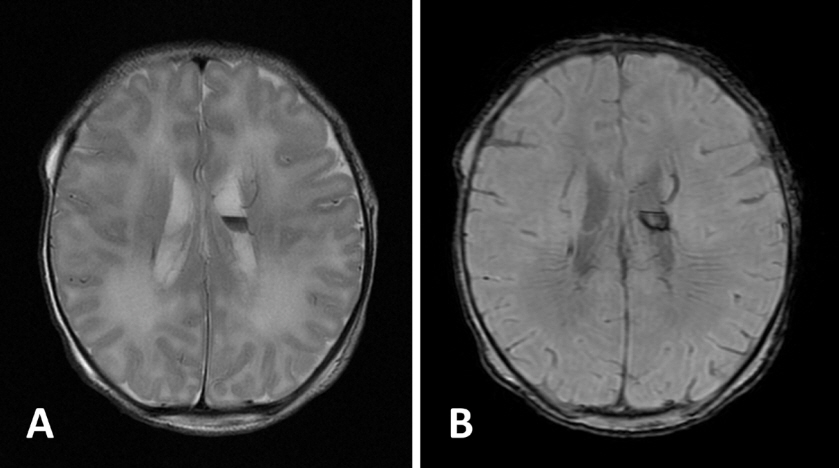
Figure 1. Brain magnetic resonance imaging of the patient with KCNQ2 encephalopathy. (A) T2-weighted imaging and (B) susceptibility-weighted imaging at day 2 of life showing cystic germinal matrix hemorrhage at the left caudothalamic groove.
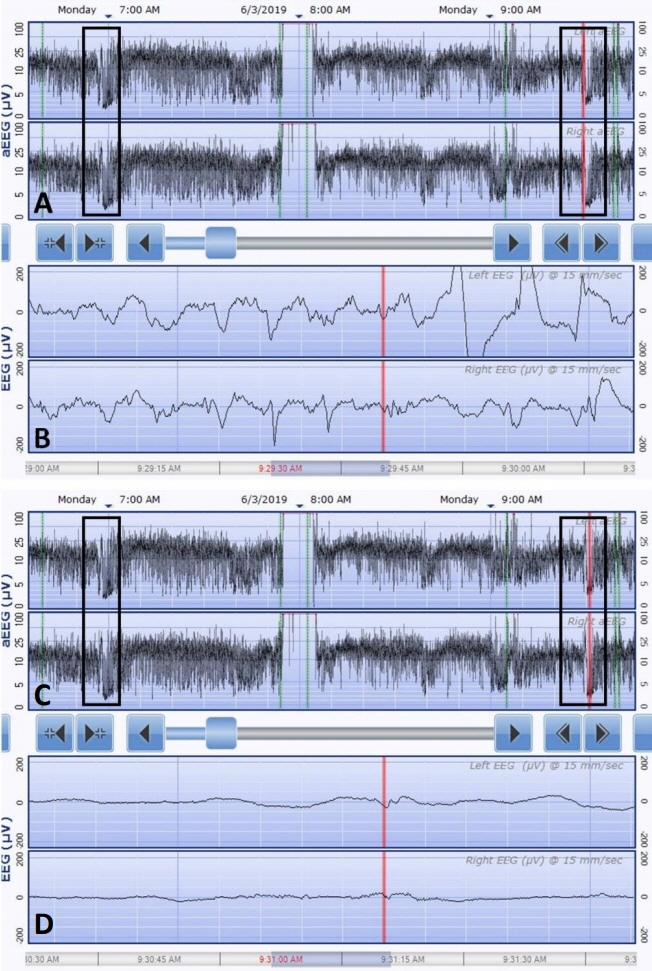
Figure 2. Seizure pattern of the patient with KCNQ2 encephalopathy on amplitude-integrated electroencephalography (aEEG). The upper panel displays the aEEG and the lower panel displays the corresponding electroencephalography (EEG) at that point indicated by the vertical red line. The aEEG shows two ictal discharges (box), a brief rise in amplitude in the upper and lower margins followed by suppression of the background pattern. The initial phase of the ictal discharges (red line in A) shows runs of spike and wave discharges on raw EEG (B). The later phase of the ictal discharges (red line in C) shows diffuse background suppression on raw EEG (D).
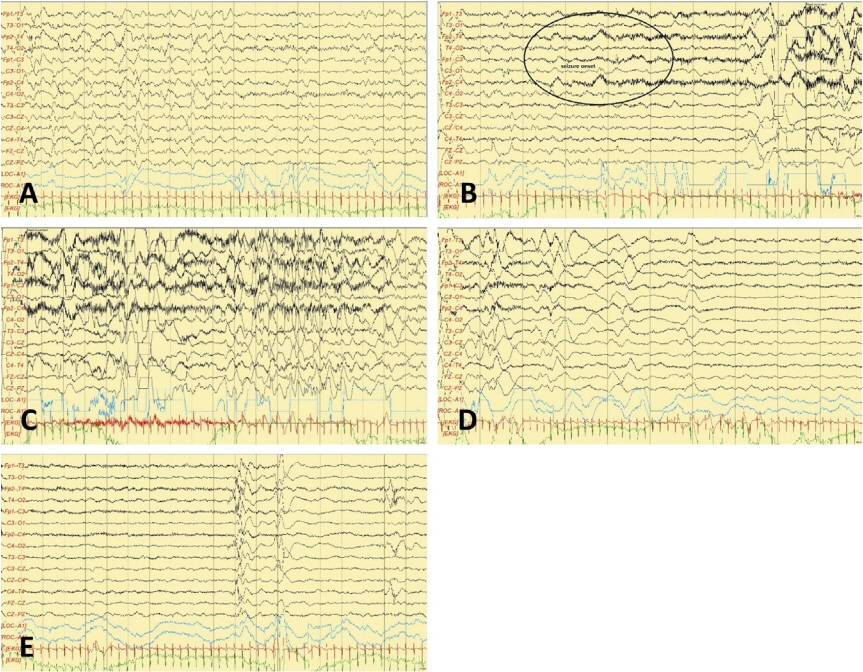
Figure 3. Seizure pattern of the patient with KCNQ2 encephalopathy on electroencephalography (EEG). (A) Interictal EEG, (B) ictal EEG shows that the fast activities began mainly from the right frontal area (circle, seizure onset), evolving to generalized spikes and waves (C) that were followed by generalized diffuse background attenuation. (D, E) Postictal EEG showing a burst suppression pattern.
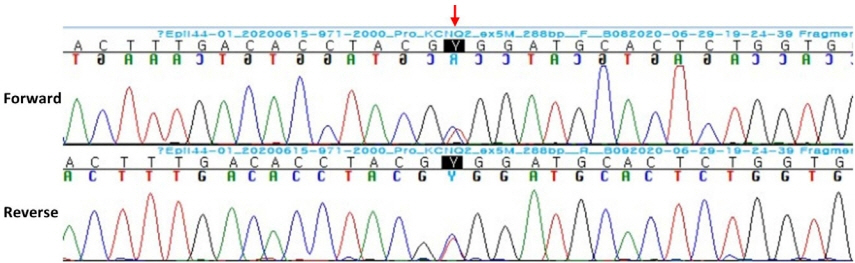
Figure 4. Sanger sequencing confirms KCNQ2 mutation [c.794C>T; p. (Ala265Val)] (arrow).
Reference
-
1. Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, et al. ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017; 58:512–21.2. Reif PS, Tsai MH, Helbig I, Rosenow F, Klein KM. Precision medicine in genetic epilepsies: break of dawn? Expert Rev Neurother. 2017; 17:381–92.3. Na JH, Shin S, Yang D, Kim B, Kim HD, Kim S, et al. Targeted gene panel sequencing in early infantile onset developmental and epileptic encephalopathy. Brain Dev. 2020; 42:438–48.4. Milh M, Boutry-Kryza N, Sutera-Sardo J, Mignot C, Auvin S, Lacoste C, et al. Similar early characteristics but variable neurological outcome of patients with a de novo mutation of KCNQ2. Orphanet J Rare Dis. 2013; 8:80.5. Biervert C, Schroeder BC, Kubisch C, Berkovic SF, Propping P, Jentsch TJ, et al. A potassium channel mutation in neonatal human epilepsy. Science. 1998; 279:403–6.6. Dedek K, Fusco L, Teloy N, Steinlein OK. Neonatal convulsions and epileptic encephalopathy in an Italian family with a missense mutation in the fifth transmembrane region of KCNQ2. Epilepsy Res. 2003; 54:21–7.7. Numis AL, Angriman M, Sullivan JE, Lewis AJ, Striano P, Nabbout R, et al. KCNQ2 encephalopathy: delineation of the electroclinical phenotype and treatment response. Neurology. 2014; 82:368–70.8. Kuersten M, Tacke M, Gerstl L, Hoelz H, Stulpnagel CV, Borggraefe I. Antiepileptic therapy approaches in KCNQ2 related epilepsy: a systematic review. Eur J Med Genet. 2020; 63:103628.9. Vilan A, Mendes Ribeiro J, Striano P, Weckhuysen S, Weeke LC, Brilstra E, et al. A distinctive ictal amplitude-integrated electroencephalography pattern in newborns with neonatal epilepsy associated with KCNQ2 mutations. Neonatology. 2017; 112:387–93.10. Sands TT, Balestri M, Bellini G, Mulkey SB, Danhaive O, Bakken EH, et al. Rapid and safe response to low-dose carbamazepine in neonatal epilepsy. Epilepsia. 2016; 57:2019–30.11. Shbarou R, Mikati MA. The expanding clinical spectrum of genetic pediatric epileptic encephalopathies. Semin Pediatr Neurol. 2016; 23:134–42.12. Ambrosio AF, Silva AP, Malva JO, Soares-da-Silva P, Carvalho AP, Carvalho CM. Carbamazepine inhibits L-type Ca2+ channels in cultured rat hippocampal neurons stimulated with glutamate receptor agonists. Neuropharmacology. 1999; 38:1349–59.13. Nguyen HM, Miyazaki H, Hoshi N, Smith BJ, Nukina N, Goldin AL, et al. Modulation of voltage-gated K+ channels by the sodium channel β1 subunit. Proc Natl Acad Sci U S A. 2012; 109:18577–82.14. de Vries LS, Hellstrom-Westas L. Role of cerebral function monitoring in the newborn. Arch Dis Child Fetal Neonatal Ed. 2005; 90:F201–7.
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Differential Findings of Interictal and Ictal Scalp Electroencephalographic Pattern between Mesial and Neocortical Temporal Lobe Epilepsies
- Frequently Identified Genetic Developmental and Epileptic Encephalopathy: A Review Focusing on Precision Medicine
- Differential Findings of Ictal EEG Pattern between Mesial and Neocortical Temporal lobe Epilepsies
- Intracranial Ictal Electroencephalographic Patterns in Parietal Lobe Epileptic Seizures : Correlation with Surgical Outcome
- Comparison of Cheek Electrode with Sphenoidal Electrodes for Identification of Ictal Onset Activity
