Ann Pediatr Endocrinol Metab.
2020 Mar;25(1):46-51. 10.6065/apem.2020.25.1.46.
Nonclassic congenital lipoid adrenal hyperplasia diagnosed at 17 months in a Korean boy with normal male genitalia: emphasis on pigmentation as a diagnostic clue
- Affiliations
-
- 1Department of Pediatrics, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea
- 2GC Genome, Yongin, Korea
- KMID: 2501037
- DOI: http://doi.org/10.6065/apem.2020.25.1.46
Abstract
- Congenital lipoid adrenal hyperplasia (CLAH) is one of the most fatal conditions caused by an abnormality of adrenal and gonadal steroidogenesis. CLAH results from loss-of-function mutations of the steroidogenic acute regulatory (STAR) gene; the disease manifests with electrolyte imbalances and hyperpigmentation in neonates or young infants due to adrenocortical hormone deficiencies, and 46, XY genetic male CLAH patients can be phenotypically female. Meanwhile, some patients with STAR mutations develop hyperpigmentation and mild signs of adrenal insufficiency, such as hypoglycemia, after infancy. These patients are classified as having nonclassic CLAH (NCCLAH) caused by STAR mutations that retain partial activity of STAR. We present the case of a Korean boy with normal genitalia who was diagnosed with NCCLAH. He presented with whole-body hyperpigmentation and electrolyte abnormalities, which were noted at the age of 17 months after an episode of sepsis with peritonitis. The compound heterozygous mutations p.Gly221Ser and c.653C>T in STAR were identified by targeted gene-panel sequencing. Skin hyperpigmentation should be considered an important clue for diagnosing NCCLAH.
Keyword
Figure
-

Fig. 1. Sanger sequencing confirmation of compound heterozygous mutations for c.653C>T and c.661G>A (p.Gly221Ser) in exon 6 of the STAR gene. Each heterozygous variant was found in the patient’s father and mother, respectively.
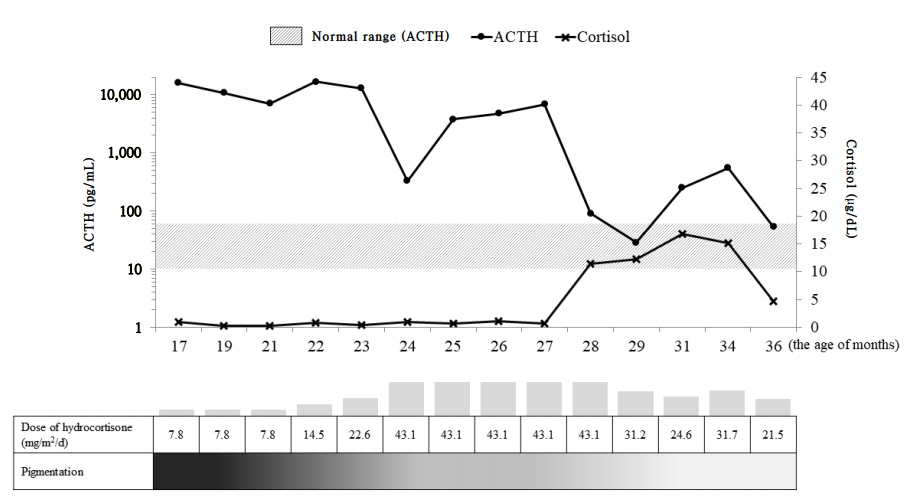
Fig. 2. Plasma adrenocorticotropic hormone (ACTH), cortisol, and pigmentation changes during hydrocortisone therapy. ACTH levels are represented by logarithm values.
Reference
-
References
1. Lin D, Sugawara T, Strauss JF 3rd, Clark BJ, Stocco DM, Saenger P, et al. Role of steroidogenic acute regulatory protein in adrenal and gonadal steroidogenesis. Science. 1995; 267:1828–31.
Article2. Bose HS, Sugawara T, Strauss JF 3rd, Miller WL; International Congenital Lipoid Adrenal Hyperplasia Consortium. The pathophysiology and genetics of congenital lipoid adrenal hyperplasia. N Engl J Med. 1996; 335:1870–8.
Article3. Kim CJ, Lin L, Huang N, Quigley CA, AvRuskin TW, Achermann JC, et al. Severe combined adrenal and gonadal deficiency caused by novel mutations in the cholesterol side chain cleavage enzyme, P450scc. J Clin Endocrinol Metab. 2008; 93:696–702.
Article4. Baker BY, Lin L, Kim CJ, Raza J, Smith CP, Miller WL, et al. Nonclassic congenital lipoid adrenal hyperplasia: a new disorder of the steroidogenic acute regulatory protein with very late presentation and normal male genitalia. J Clin Endocrinol Metab. 2006; 91:4781–5.
Article5. Nakae J, Tajima T, Sugawara T, Arakane F, Hanaki K, Hotsubo T, et al. Analysis of the steroidogenic acute regulatory protein (StAR) gene in Japanese patients with congenital lipoid adrenal hyperplasia. Hum Mol Genet. 1997; 6:571–6.
Article6. Flück CE, Pandey AV, Dick B, Camats N, Fernández-Cancio M, Clemente M, et al. Characterization of novel StAR (steroidogenic acute regulatory protein) mutations causing non-classic lipoid adrenal hyperplasia. PLoS One. 2011; 6:e20178.
Article7. Kim JM, Choi JH, Lee JH, Kim GH, Lee BH, Kim HS, et al. High allele frequency of the p.Q258X mutation and identification of a novel mis-splicing mutation in the STAR gene in Korean patients with congenital lipoid adrenal hyperplasia. Eur J Endocrinol. 2011; 165:771–8.
Article8. Gucev ZS, Tee MK, Chitayat D, Wherrett DK, Miller WL. Distinguishing deficiencies in the steroidogenic acute regulatory protein and the cholesterol side chain cleavage enzyme causing neonatal adrenal failure. J Pediatr. 2013; 162:819–22.
Article9. Rubtsov P, Karmanov M, Sverdlova P, Spirin P, Tiulpakov A. A novel homozygous mutation in CYP11A1 gene is associated with late-onset adrenal insufficiency and hypospadias in a 46,XY patient. J Clin Endocrinol Metab. 2009; 94:936–9.10. Chen X, Baker BY, Abduljabbar MA, Miller WL. A genetic isolate of congenital lipoid adrenal hyperplasia with atypical clinical findings. J Clin Endocrinol Metab. 2005; 90:835–40.
Article11. Sahakitrungruang T. Clinical and molecular review of atypical congenital adrenal hyperplasia. Ann Pediatr Endocrinol Metab. 2015; 20:1–7.
Article12. Metherell LA, Naville D, Halaby G, Begeot M, Huebner A, Nürnberg G, et al. Nonclassic lipoid congenital adrenal hyperplasia masquerading as familial glucocorticoid deficiency. J Clin Endocrinol Metab. 2009; 94:3865–71.
Article13. Burget L, Parera LA, Fernandez-Cancio M, Gräni R, Henzen C, Flück CE. A rare cause of primary adrenal insufficiency due to a homozygous Arg188Cys mutation in the STAR gene. Endocrinol Diabetes Metab Case Rep. 2018; 2018.
Article14. Sahakitrungruang T, Soccio RE, Lang-Muritano M, Walker JM, Achermann JC, Miller WL. Clinical, genetic, and functional characterization of four patients carrying partial loss-of-function mutations in the steroidogenic acute regulatory protein (StAR). J Clin Endocrinol Metab. 2010; 95:3352–9.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Congenital Lipoid Adrenal Hyperplasia Developed in a Brother and Sister
- A Case of Congenital Lipoid Adrenal Hyperplasia
- A Case of Congenital Lipoid Adrenal Hyperplasia: Early Diagnosis by Using Computed Tomography
- Congenital lipoid adrenal hyperplasia
- Two Cases of Secondary Central Precocious Puberty Occurred in Congenital Adrenal Hyperplasia
