Severe or Profound Sensorineural Hearing Loss Caused by Novel USH2A Variants in Korea: Potential Genotype-Phenotype Correlation
- Affiliations
-
- 1Department of Otorhinolaryngology-Head and Neck Surgery, Seoul National University Bundang Hospital, Seongnam, Korea
- 2Department of Ophthalmology, Seoul National University Bundang Hospital, Seoul National University College of Medicine, Seongnam, Korea
- KMID: 2500283
- DOI: http://doi.org/10.21053/ceo.2019.00990
Abstract
Objectives
. We, herein, report two novel USH2A variants from two unrelated Korean families and their clinical phenotypes, with attention to severe or more than severe sensorineural hearing loss (SNHL).
Methods
. Two postlingually deafened subjects (SB237-461, M/46 and SB354-692, F/34) with more than severe SNHL and also with suspicion of Usher syndrome type II (USH2) were enrolled. A comprehensive audiological and ophthalmological assessments were evaluated. We conducted the whole exome sequencing and subsequent pathogenicity prediction analysis.
Results
. We identified the following variants of USH2A from the two probands manifesting more than severe SNHL and retinitis pigmentosa (RP): compound heterozygosity for a nonsense (c.8176C>T: p.R2723X) and a missense variant (c.1823G>A: p.C608Y) in SB237, and compound heterozygosity for two frameshift variants (c.14835delT: p.S4945fs & c.13112_13115delAAAT: p.G4371fs) in SB354. Based on the American College of Medical Genetics and Genomics/Association for Molecular Pathology guidelines, two novel variants, c.1823G>A: p.C608Y and c.14835delT: p.Ser4945fs, can be classified as “uncertain significance” and “pathogenic,” respectively. The audiogram exhibited more than severe SNHL and a down-sloping configuration, necessitating cochlear implantation. The ophthalmic examinations revealed typical features of RP. Interestingly, one proband (SB 354-692) carrying two truncating compound heterozygous variants exhibited more severe hearing loss than the other proband (SB 237-461), carrying one truncation with one missense variant.
Conclusion
. Our results provide insight on the expansion of audiological spectrum encompassing more than severe SNHL in Korean subjects harboring USH2A variants, suggesting that USH2A should also be included in the candidate gene of cochlear implantation. A specific combination of USH2A variants causing truncating proteins in both alleles could demonstrate more severe audiological phenotype than that of USH2A variants carrying one truncating mutation and one missense mutation, suggesting a possible genotype-phenotype correlation. The understanding of audiological complexity associated with USH2A will be helpful for genetic counseling and treatment starategy.
Keyword
Figure
-
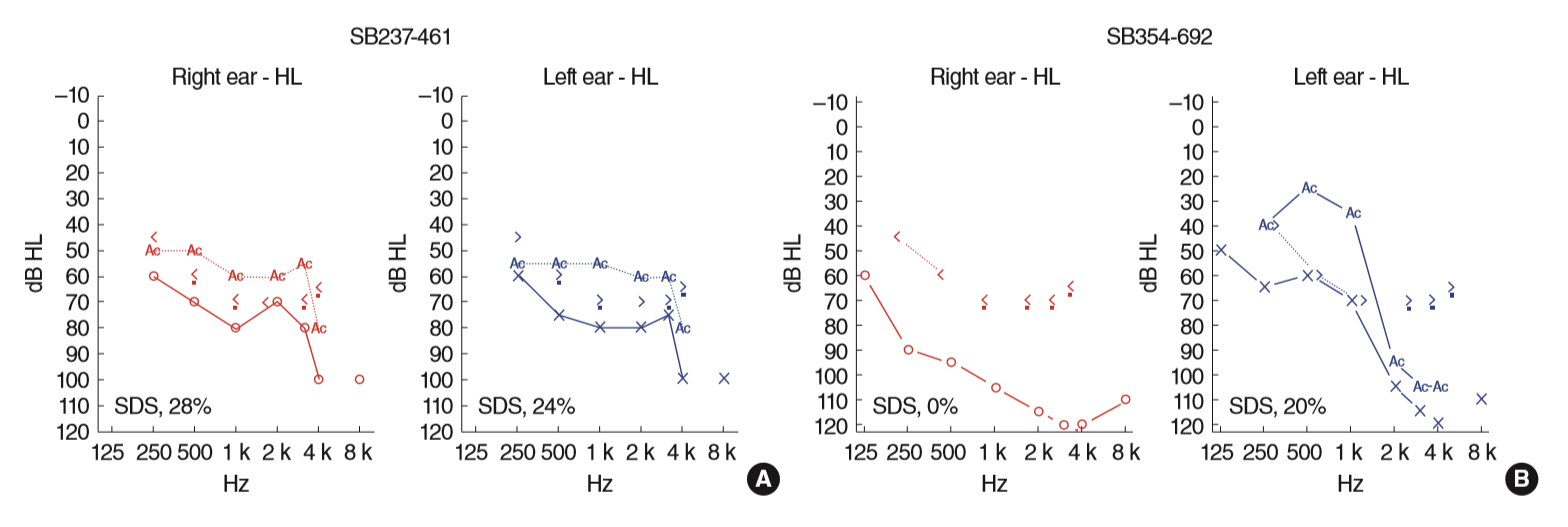
Fig. 1. Audiological phenotypes of affected subjects with Usher syndrome type II (USH2) variants. (A) SB237-461, bilateral severe down-sloping sensorineural hearing loss (SNHL). (B) SB354-692, profound down-sloping SNHL on the right ear and severe down-sloping SNHL on the left ear. Red and blue lines represent the right and left ear hearing thresholds, respectively. Numbers with a percentage demonstrate the speech audiometry score in each evaluated ear. SDS, speech discrimination score.
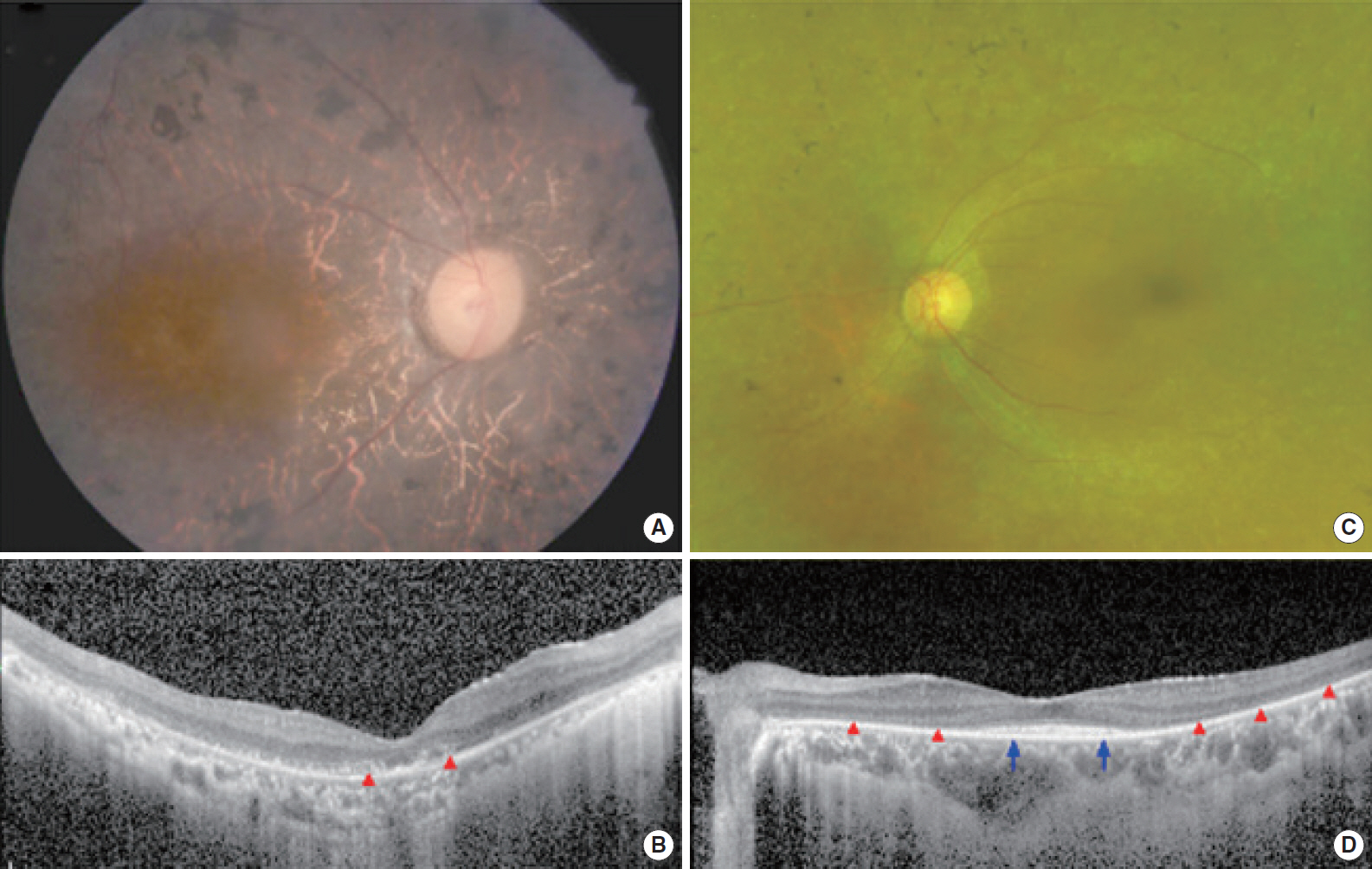
Fig. 2. Representative color fundus photographs and optical coherence tomography (OCT) images for two patients with typical features of retinitis pigmentosa. (A) A right fundus image from patient SB237-461 exhibits diffuse retinal pigmentary changes, vascular attenuation and waxy disc pallor. (B) An ipsilateral spectral-domain OCT image shows the diffuse disruption of photoreceptors (red arrowheads) in the macula. (C) A left fundus image from patient SB354-692 shows diffuse retinal degeneration but relatively preserved macula. (D) An OCT image shows the preservation of photoreceptors in fovea (blue arrows) whereas extrafoveal photoreceptors disappeared (red arrowheads).

Fig. 3. Sanger sequencing results of SB237 and SB354, as well as the conservation of residue p.R2723X and p.C608Y from a various species. (A) SB237: candidate variants of USH2A with recessive inheritance (c.8167C>T: p.R2723X and c.1823G>A: p.C608Y) on the basis of Sanger sequencing chromatograms. (B) The residues p.R2723X and p.C608Y were well conserved in known USH2A orthologs across different species. (C) SB354: candidate variants of USH2A with recessive inheritance (c.14835delT: p.S4945fs and c.13112_13115delAAAT: p.G4371fs) according to Sanger sequencing chromatograms. Het, heterozygous.
Reference
-
1. van Wijk E, Pennings RJ, te Brinke H, Claassen A, Yntema HG, Hoefsloot LH, et al. Identification of 51 novel exons of the Usher syndrome type 2A (USH2A) gene that encode multiple conserved functional domains and that are mutated in patients with Usher syndrome type II. Am J Hum Genet. 2004; Apr. 74(4):738–44.
Article2. Smith RJ, Berlin CI, Hejtmancik JF, Keats BJ, Kimberling WJ, Lewis RA, et al. Clinical diagnosis of the Usher syndrome: Usher Syndrome Consortium. Am J Med Genet. 1994; Mar. 50(1):32–8.3. Ahmed ZM, Riazuddin S, Riazuddin S, Wilcox ER. The molecular genetics of Usher syndrome. Clin Genet. 2003; Jun. 63(6):431–44.
Article4. Otterstedde CR, Spandau U, Blankenagel A, Kimberling WJ, Reisser C. A new clinical classification for Usher’s syndrome based on a new subtype of Usher’s syndrome type I. Laryngoscope. 2001; Jan. 111(1):84–6.
Article5. Spandau UH, Rohrschneider K. Prevalence and geographical distribution of Usher syndrome in Germany. Graefes Arch Clin Exp Ophthalmol. 2002; Jun. 240(6):495–8.
Article6. Weston MD, Luijendijk MW, Humphrey KD, Moller C, Kimberling WJ. Mutations in the VLGR1 gene implicate G-protein signaling in the pathogenesis of Usher syndrome type II. Am J Hum Genet. 2004; Feb. 74(2):357–66.
Article7. van Wijk E, van der Zwaag B, Peters T, Zimmermann U, Te Brinke H, Kersten FF, et al. The DFNB31 gene product whirlin connects to the Usher protein network in the cochlea and retina by direct association with USH2A and VLGR1. Hum Mol Genet. 2006; Mar. 15(5):751–65.
Article8. Booth KT, Azaiez H, Kahrizi K, Simpson AC, Tollefson WT, Sloan CM, et al. PDZD7 and hearing loss: More than just a modifier. Am J Med Genet A. 2015; Dec. 167(12):2957–65.9. Chen Q, Zou J, Shen Z, Zhang W, Yang J. Whirlin and PDZ domaincontaining 7 (PDZD7) proteins are both required to form the quaternary protein complex associated with Usher syndrome type 2. J Biol Chem. 2014; Dec. 289(52):36070–88.
Article10. Bhattacharya G, Miller C, Kimberling WJ, Jablonski MM, Cosgrove D. Localization and expression of Usherin: a novel basement membrane protein defective in people with Usher’s syndrome type IIa. Hear Res. 2002; Jan. 163(1-2):1–11.
Article11. Liu X, Bulgakov OV, Darrow KN, Pawlyk B, Adamian M, Liberman MC, et al. Usherin is required for maintenance of retinal photoreceptors and normal development of cochlear hair cells. Proc Natl Acad Sci U S A. 2007; Mar. 104(11):4413–8.
Article12. Pearsall N, Bhattacharya G, Wisecarver J, Adams J, Cosgrove D, Kimberling W. Usherin expression is highly conserved in mouse and human tissues. Hear Res. 2002; Dec. 174(1-2):55–63.
Article13. Chen X, Sheng X, Liu X, Li H, Liu Y, Rong W, et al. Targeted next-generation sequencing reveals novel USH2A mutations associated with diverse disease phenotypes: implications for clinical and molecular diagnosis. PLoS One. 2014; Aug. 9(8):e105439.
Article14. Lenassi E, Vincent A, Li Z, Saihan Z, Coffey AJ, Steele-Stallard HB, et al. A detailed clinical and molecular survey of subjects with nonsyndromic USH2A retinopathy reveals an allelic hierarchy of disease-causing variants. Eur J Hum Genet. 2015; Oct. 23(10):1318–27.
Article15. McGee TL, Seyedahmadi BJ, Sweeney MO, Dryja TP, Berson EL. Novel mutations in the long isoform of the USH2A gene in patients with Usher syndrome type II or non-syndromic retinitis pigmentosa. J Med Genet. 2010; Jul. 47(7):499–506.
Article16. Dreyer B, Brox V, Tranebjaerg L, Rosenberg T, Sadeghi AM, Moller C, et al. Spectrum of USH2A mutations in Scandinavian patients with Usher syndrome type II. Hum Mutat. 2008; Mar. 29(3):451.
Article17. Pennings RJ, Huygen PL, Weston MD, van Aarem A, Wagenaar M, Kimberling WJ, et al. Pure tone hearing thresholds and speech recognition scores in Dutch patients carrying mutations in the USH2A gene. Otol Neurotol. 2003; Jan. 24(1):58–63.
Article18. Sadeghi AM, Cohn ES, Kimberling WJ, Halvarsson G, Moller C. Expressivity of hearing loss in cases with Usher syndrome type IIA. Int J Audiol. 2013; Dec. 52(12):832–7.
Article19. Hartel BP, Lofgren M, Huygen PL, Guchelaar I, Lo-A-Njoe Kort N, Sadeghi AM, et al. A combination of two truncating mutations in USH2A causes more severe and progressive hearing impairment in Usher syndrome type IIa. Hear Res. 2016; Sep. 339:60–8.
Article20. Lee SY, Nam DW, Koo JW, De Ridder D, Vanneste S, Song JJ. No auditory experience, no tinnitus: lessons from subjects with congenital- and acquired single-sided deafness. Hear Res. 2017; Oct. 354:9–15.21. Kim NK, Kim AR, Park KT, Kim SY, Kim MY, Nam JY, et al. Whole-exome sequencing reveals diverse modes of inheritance in sporadic mild to moderate sensorineural hearing loss in a pediatric population. Genet Med. 2015; Nov. 17(11):901–11.
Article22. Kim BJ, Han JH, Park HR, Kim MY, Kim AR, Oh SH, et al. A clinical guidance to DFNA22 drawn from a Korean cohort study with an autosomal dominant deaf population: a retrospective cohort study. J Gene Med. 2018; Jun. 20(6):e3019.
Article23. Jaijo T, Aller E, Garcia-Garcia G, Aparisi MJ, Bernal S, Avila-Fernandez A, et al. Microarray-based mutation analysis of 183 Spanish families with Usher syndrome. Invest Ophthalmol Vis Sci. 2010; Mar. 51(3):1311–7.
Article24. Sengillo JD, Cabral T, Schuerch K, Duong J, Lee W, Boudreault K, et al. Electroretinography reveals difference in cone function between syndromic and nonsyndromic USH2A patients. Sci Rep. 2017; Sep. 7(1):11170.
Article25. Oza AM, DiStefano MT, Hemphill SE, Cushman BJ, Grant AR, Siegert RK, et al. Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum Mutat. 2018; Nov. 39(11):1593–613.
Article26. Abou Tayoun AN, Pesaran T, DiStefano MT, Oza A, Rehm HL, Biesecker LG, et al. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum Mutat. 2018; Nov. 39(11):1517–24.
Article27. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015; May. 17(5):405–24.
Article28. Aller E, Najera C, Millan JM, Oltra JS, Perez-Garrigues H, Vilela C, et al. Genetic analysis of 2299delG and C759F mutations (USH2A) in patients with visual and/or auditory impairments. Eur J Hum Genet. 2004; May. 12(5):407–10.
Article29. Liu XZ, Hope C, Liang CY, Zou JM, Xu LR, Cole T, et al. A mutation (2314delG) in the Usher syndrome type IIA gene: high prevalence and phenotypic variation. Am J Hum Genet. 1999; Apr. 64(4):1221–5.
Article30. Schultz JM, Bhatti R, Madeo AC, Turriff A, Muskett JA, Zalewski CK, et al. Allelic hierarchy of CDH23 mutations causing non-syndromic deafness DFNB12 or Usher syndrome USH1D in compound heterozygotes. J Med Genet. 2011; Nov. 48(11):767–75.
Article31. Blanco-Kelly F, Jaijo T, Aller E, Avila-Fernandez A, Lopez-Molina MI, Gimenez A, et al. Clinical aspects of Usher syndrome and the USH2A gene in a cohort of 433 patients. JAMA Ophthalmol. 2015; Feb. 133(2):157–64.32. Abadie C, Blanchet C, Baux D, Larrieu L, Besnard T, Ravel P, et al. Audiological findings in 100 USH2 patients. Clin Genet. 2012; Nov. 82(5):433–8.
Article33. Bernal S, Meda C, Solans T, Ayuso C, Garcia-Sandoval B, Valverde D, et al. Clinical and genetic studies in Spanish patients with Usher syndrome type II: description of new mutations and evidence for a lack of genotype--phenotype correlation. Clin Genet. 2005; Sep. 68(3):204–14.
Article34. Dai H, Zhang X, Zhao X, Deng T, Dong B, Wang J, et al. Identification of five novel mutations in the long isoform of the USH2A gene in Chinese families with Usher syndrome type II. Mol Vis. 2008; 14:2067–75.35. Kim BJ, Kim AR, Lee C, Kim SY, Kim NK, Chang MY, et al. Discovery of CDH23 as a significant contributor to progressive postlingual sensorineural hearing loss in Koreans. PLoS One. 2016; Oct. 11(10):e0165680.
Article36. Nagase Y, Kurata K, Hosono K, Suto K, Hikoya A, Nakanishi H, et al. Visual outcomes in Japanese patients with retinitis pigmentosa and usher syndrome caused by USH2A mutations. Semin Ophthalmol. 2018; 33(4):560–5.
Article37. Pierrache LH, Hartel BP, van Wijk E, Meester-Smoor MA, Cremers FP, de Baere E, et al. Visual prognosis in USH2A-associated retinitis pigmentosa is worse for patients with usher syndrome type IIa than for those with nonsyndromic retinitis pigmentosa. Ophthalmology. 2016; May. 123(5):1151–60.
Article38. Ng TK, Tang W, Cao Y, Chen S, Zheng Y, Xiao X, et al. Whole exome sequencing identifies novel USH2A mutations and confirms Usher syndrome 2 diagnosis in Chinese retinitis pigmentosa patients. Sci Rep. 2019; Apr. 9(1):5628.
Article39. Hartel BP, van Nierop JW, Huinck WJ, Rotteveel LJ, Mylanus EA, Snik AF, et al. Cochlear implantation in patients with usher syndrome type IIa increases performance and quality of life. Otol Neurotol. 2017; Jul. 38(6):e120–7.
Article40. Neveling K, Collin RW, Gilissen C, van Huet RA, Visser L, Kwint MP, et al. Next-generation genetic testing for retinitis pigmentosa. Hum Mutat. 2012; Jun. 33(6):963–72.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- A Case of Bilateral Cochlear Implants in Bilateral Profound Sudden Sensorineural Hearing Loss After COVID-19 Infection
- Strategy for the Customized Mass Screening of Genetic Sensorineural Hearing Loss in Koreans
- Outcomes of Severe to Profound Idiopathic Sudden Sensorineural Hearing Loss
- Update on the Management for Sensorineural Hearing Loss in Children
- A Case of Cochlear Implantation after Bilateral Temporal Bone Fracture
