Pediatr Gastroenterol Hepatol Nutr.
2014 Dec;17(4):239-247. 10.5223/pghn.2014.17.4.239.
Does Type I Truly Dominate Hepatic Glycogen Storage Diseases in Korea?: A Single Center Study
- Affiliations
-
- 1Department of Pediatrics, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea. i101016@skku.edu
- 2Department of Laboratory Medicine and Genetics, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea.
- KMID: 2315498
- DOI: http://doi.org/10.5223/pghn.2014.17.4.239
Abstract
- PURPOSE
There are no studies of hepatic glycogen storage diseases (GSDs) other than type I and III in Korea. We aimed on investigating the characteristics of hepatic GSDs in Korea diagnosed and followed at a single center.
METHODS
We retrospectively analyzed patients who were diagnosed as GSD and followed at Samsung Medical Center from January, 1997 to December, 2013. Clinical manifestations, laboratory results, treatment, and prognosis were investigated.
RESULTS
Twenty-one patients were included in the study. The types of 17 patients were confirmed by enzyme activity tests and/or gene analysis. GSD Ia was diagnosed in 7 patients (33.3%), Ib in 1 patient (4.8%), III in 2 patients (9.5%), IV in 1 patient (4.8%), and IX in 6 patients (28.6%). Types other than GSD I constituted 52.9% (9/17) of the patients diagnosed with a specific type of hepatic GSD. The median age at presentation was 2 years. Hepatomegaly was observed in 95.2%, elevated liver transaminases in 90.5%, and hyperlactacidemia in 81.0% of the patients. The duration for follow-up was 77+/-62.0 months. Uncooked corn starch was initiated in all the patients. No mortality was observed during the follow-up period, and liver transplantation was performed in 14.3%.
CONCLUSION
Types other than GSD I comprised more than half of the patients diagnosed with a specific type of hepatic GSD. Clinical suspicion and thorough evaluation of hepatic GSDs in Korea should be focused not only on GSD I, but also on other types.
Keyword
MeSH Terms
Figure
-
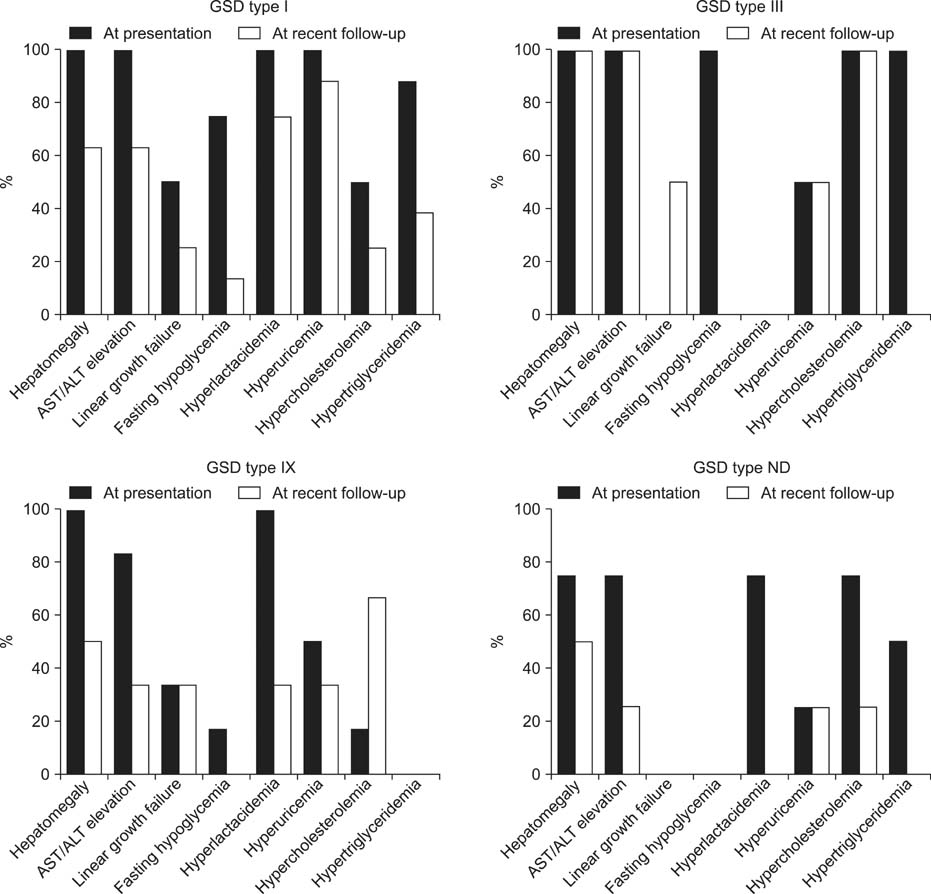
Fig. 1 Clinicolaboratory findings of hepatic glycogen strorage diseases at presentation and at recent follow up. GSD: glycogen storage disease; AST/ALT: aspartate aminotransferase/alanine aminotransferase; ND: not determined.
Reference
-
1. Applegarth DA, Toone JR, Lowry RB. Incidence of inborn errors of metabolism in British Columbia, 1969-1996. Pediatrics. 2000; 105:e10.
Article2. Ozen H. Glycogen storage diseases: new perspectives. World J Gastroenterol. 2007; 13:2541–2553.
Article3. Chen YT. Glycogen storage diseases. In : Scriver CR, Beaudet AL, Sly WS, Vale D, Childs B, Kinzler KW, editors. The metabolic & molecular basis of inherited diseases. 8th ed. New York: McGraw-Hill;2001. p. 1521–1552.4. Froissart R, Piraud M, Boudjemline AM, Vianey-Saban C, Petit F, Hubert-Buron A, et al. Glucose-6-phosphatase deficiency. Orphanet J Rare Dis. 2011; 6:27.
Article5. Wolfsdorf JI, Weinstein DA. Glycogen storage diseases. Rev Endocr Metab Disord. 2003; 4:95–102.
Article6. Chen YT, Cornblath M, Sidbury JB. Cornstarch therapy in type I glycogen-storage disease. N Engl J Med. 1984; 310:171–175.
Article7. Rake JP, Visser G, Labrune P, Leonard JV, Ullrich K, Smit GP. European Study on Glycogen Storage Disease Type I (ESGSD I). Guidelines for management of glycogen storage disease type I - European Study on Glycogen Storage Disease Type I (ESGSD I). Eur J Pediatr. 2002; 161:Suppl 1. S112–S119.8. Matern D, Starzl TE, Arnaout W, Barnard J, Bynon JS, Dhawan A, et al. Liver transplantation for glycogen storage disease types I, III, and IV. Eur J Pediatr. 1999; 158:Suppl 2. S43–S48.
Article9. Iyer SG, Chen CL, Wang CC, Wang SH, Concejero AM, Liu YW, et al. Long-term results of living donor liver transplantation for glycogen storage disorders in children. Liver Transpl. 2007; 13:848–852.
Article10. Willems PJ, Gerver WJ, Berger R, Fernandes J. The natural history of liver glycogenosis due to phosphorylase kinase deficiency: a longitudinal study of 41 patients. Eur J Pediatr. 1990; 149:268–271.
Article11. Smit GP, Fernandes J, Leonard JV, Matthews EE, Moses SW, Odievre M, et al. The long-term outcome of patients with glycogen storage diseases. J Inherit Metab Dis. 1990; 13:411–418.
Article12. Rake JP, Visser G, Labrune P, Leonard JV, Ullrich K, Smit GP. Glycogen storage disease type I: diagnosis, management, clinical course and outcome. Results of the European Study on Glycogen Storage Disease Type I (ESGSD I). Eur J Pediatr. 2002; 161:Suppl 1. S20–S34.
Article13. Kido J, Nakamura K, Matsumoto S, Mitsubuchi H, Ohura T, Shigematsu Y, et al. Current status of hepatic glycogen storage disease in Japan: clinical manifestations, treatments and long-term outcomes. J Hum Genet. 2013; 58:285–292.
Article14. Hershkovitz E, Forschner I, Mandel H, Spiegel R, Lerman-Sagie T, Anikster Y, et al. Glycogen storage disease type III in Israel: presentation and long-term outcome. Pediatr Endocrinol Rev. 2014; 11:318–323.15. Lee SY, Seo JK. Uncooked cornstarch therapy in type I glycogen-storage disease (GSD-I). J Korean Pediatr Soc. 1995; 38:36–46.16. Kim JW, Park JY, Seo JK. Mutation analysis of Korean patients with glycogen storage disease type Ia. Korean J Pediatr Gastroenterol Nutr. 2001; 4:213–217.
Article17. Yang HR, Seo JK. Long-term outcome of glycogen storage disease type 1; analysis of risk factors for hepatic adenoma. Korean J Pediatr Gastroenterol Nutr. 2003; 6:129–139.
Article18. Choi J, Ko JM, Kim GH, Yoo HW. Clinical manifestation and effect of corn starch on height growth in Korean patients with glycogen storage disease type Ia. J Korean Soc Pediatr Endocrinol. 2007; 12:35–40.19. Ko JM, Kim GH, Yoo HW. AGL gene mutation and clinical features in Korean patients with glycogen storage disease type III. J Genet Med. 2007; 4:72–79.20. Demo E, Frush D, Gottfried M, Koepke J, Boney A, Bali D, et al. Glycogen storage disease type III-hepatocellular carcinoma a long-term complication? J Hepatol. 2007; 46:492–498.
Article21. Hicks J, Wartchow E, Mierau G. Glycogen storage diseases: a brief review and update on clinical features, genetic abnormalities, pathologic features, and treatment. Ultrastruct Pathol. 2011; 35:183–196.
Article22. Coleman RA, Winter HS, Wolf B, Chen YT. Glycogen debranching enzyme deficiency: long-term study of serum enzyme activities and clinical features. J Inherit Metab Dis. 1992; 15:869–881.
Article23. Talente GM, Coleman RA, Alter C, Baker L, Brown BI, Cannon RA, et al. Glycogen storage disease in adults. Ann Intern Med. 1994; 120:218–226.
Article24. L'herminé-Coulomb A, Beuzen F, Bouvier R, Rolland MO, Froissart R, Menez F, et al. Fetal type IV glycogen storage disease: clinical, enzymatic, and genetic data of a pure muscular form with variable and early antenatal manifestations in the same family. Am J Med Genet A. 2005; 139A:118–122.25. Moses SW, Parvari R. The variable presentations of glycogen storage disease type IV: a review of clinical, enzymatic and molecular studies. Curr Mol Med. 2002; 2:177–188.
Article26. Hidaka F, Sawada H, Matsuyama M, Nunoi H. A novel mutation of the PHKA2 gene in a patient with X-linked liver glycogenosis type 1. Pediatr Int. 2005; 47:687–690.
Article27. Davidson JJ, Ozçelik T, Hamacher C, Willems PJ, Francke U, Kilimann MW. cDNA cloning of a liver isoform of the phosphorylase kinase alpha subunit and mapping of the gene to Xp22.2-p22.1, the region of human X-linked liver glycogenosis. Proc Natl Acad Sci U S A. 1992; 89:2096–2100.
Article28. Burwinkel B, Shiomi S, Al Zaben A, Kilimann MW. Liver glycogenosis due to phosphorylase kinase deficiency: PHKG2 gene structure and mutations associated with cirrhosis. Hum Mol Genet. 1998; 7:149–154.
Article29. Beauchamp NJ, Dalton A, Ramaswami U, Niinikoski H, Mention K, Kenny P, et al. Glycogen storage disease type IX: High variability in clinical phenotype. Mol Genet Metab. 2007; 92:88–99.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- A case of multiple hepatic adenomas and gout with glycogen storage disease type Ia
- A Case of Glycogen Storage Disease Type IIa
- A Case of Adult Onset Glycogen Storage Myopathy
- Hepatic adenomatosis in glycogen storage disease
- Long-term Outcome of Glycogen Storage Disease Type 1; Analysis of Risk Factors for Hepatic Adenoma
