A case Report of a Classic Cystic fibrosis Pediatric Patient in Korea Carrying Very Rare CFTR Gene Mutations (D993Y and Q220X)
- Affiliations
-
- 1Department of Pediatrics and Institute of Allergy, Yonsei University College of Medicine, Seoul, Korea. mhsohn@yuhs.ac
- 2Department of Pharmacology, Yonsei University College of Medicine, Seoul, Korea.
- 3Department of Diagnostic Radiology, Yonsei University College of Medicine, Seoul, Korea.
Abstract
- Cystic fibrosis is the most common autosomal recessive disease in Caucasian. Cystic fibrosis is caused by cystic fibrosis transmembrane conductance regulator (CFTR) gene mutations that lead to dysfunction of chloride ion channel regulations in the epithelium. Cystic fibrosis can affect multiple organ functions, resulting in various signs and symptoms. Typically, chronic airway infection, maldigestion, failure to thrive, and male infertility can occur. There are approximately 1800 CFTR gene mutations which have been identified thus far. However, there are only a few types of mutations reported in Korea because the prevalence of the disease is different among ethnicitiess and nations. Despite its rarity, reports of CFTR mutations or diagnosed patients on the rise. Therefore, we have to detect better outcomes as early as possible based on a precise understanding of the disease entity. We report a 9-year-old girl carrying D339Y and Q220X gene mutations, as the first case report of a D339Y mutation in Korea.
Keyword
MeSH Terms
Figure
-

Fig. 1. (A) Plain chest X-ray shows diffuse nodular densities in both lungs. (B) Water's view shows both maxillary sinusitis.
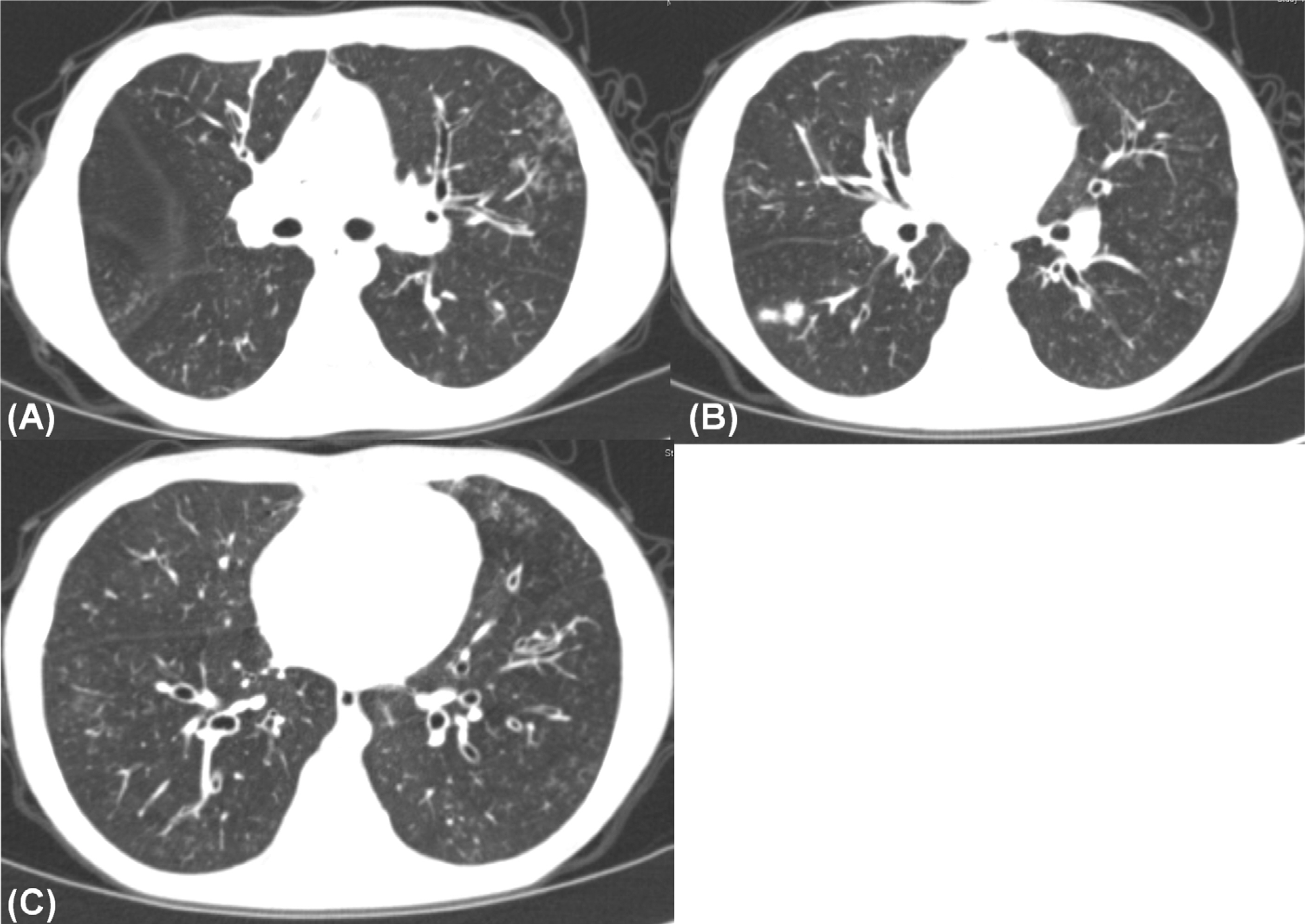
Fig. 2. Multiple tiny air-space nodules are seen in both lung fields. Also bronchiectasis in both lungs are observed on computer tomographic imaging.

Fig. 3. Denaturing gradient gel electrophoresis (DGGE) results of the patient and her family. (A) There are single nucleotide polymorphisms (SNPs) in exon 6a of M and P. (B) There are SNPs in exon 16 of F, P, B and S. Abbreviations: F, father; M, mother; P, patient; B, brother; S, sister

Fig. 4. A pedigree of the patient diagnosed as cystic fibrosis. 2 kinds of disease-causing mutations are found in this patient. Q220X mutation is from her mother, and D993Y came from her father. In the other words, her parents are unaffected carriers. She has 2 siblings possessing D993Y. Both of them are not only the unaffected but also carriers.
Reference
-
References
1. Flume PA, Stenbit A. Making the diagnosis of cystic fibrosis. Am J Med Sci. 2008; 335:51–4.
Article2. Wallis C. Diagnosis and presentation of cystic fibrosis. Chernick V, Boat T, Wilmott R, Bush A, editors. editors.Kendig's Disorders of the Respiratory tract in Children. 7th Ed.Philadelphia, Pa: Saunders Elsevier;2007. p. 866–72.
Article3. Moskowitz SM, Chimel JF, Sternen DL, Cheng E, Gibson RL, Marshall SG, et al. Clinical practice and genetic counseling for cystic fibrosis and CFTR-related disorders. Genet Med. 2008; 10:851–68.
Article4. Farrell PM, Rosenstein BJ, White TB, Accurso FJ, Castellani C, Cutting GR, et al. Guidelines for diagnosis of cystic fibrosis in newborns through older adults: Cystic Fibrosis Foundation Consensus Report. J Pediatr. 2008; 153:S4–14.
Article5. Bobadilla JL, Macek M Jr, Fine JP, Farrell PM. Cystic fibrosis: A worldwide analysis of CFTR mutations-correlation with incidence data and application to screening. Hum Mutat. 2002; 19:575–606.6. Legrys VA. Sweat testing for the diagnosis of cystic fibrosis: practical considerations. J Pediatr. 1996; 129:892–7.
Article7. Imaizumi Y. Incidence and mortality rates of cystic fibrosis in Japan, 1969–1992. Am J Med Genet. 1995 Aug 28; 58:161–8.
Article8. Yamashiro Y, Shimizu T, Oguchi S, Shioya T, Nagata S, Ohtsuka Y. The estimated incidence of cystic fibrosis in Japan. J Pediatr Gastroenterol Nur. 1997; 24:544–7.
Article9. Li N, Pei P, Bu DF, He B, Wang GF. A novel CFTR mutation found in a Chinese patient with cystic fibrosis. Chin Med J. 2006; 119:103–9.
Article10. Julian zielenski. Geneotype and phenotype in cystic fibrosis. Respiration. 2000; 67:117–33.11. The Cystic fibrosis Genetic Analysis Consortium. Cystic Fibrosis Mutation Database. http://www.genet.sickkids.on.ca/cftr.12. Lee JH, Choi JH, Namkung W, Hanrahan JW, Chang J, Song SY, et al. A haplotype-based molecular analysis of CFTR mutations associated with respiratory and pancreatic disease. Hum Mol Genet. 2003; 12:2321–32.13. Shackleton S, Hull J, Dear S, Seller A, Thomson A, Harris A. Identification of rare and novel mutations in the CFTR genes of CF patients in Southern England. Hum Mutat. 1994; 3:141–51.
Article14. Koh WJ, Ki CS, Kim JW, Kim JH, Lim SY. Report of a Korean patient with cystic fibrosis, carrying Q98R and Q220X mutations in the CFTR gene. J Korean Med Sci. 2006; 21:563–6.
Article15. Moon HR, Ko TS, Ko YY, Choi JH, Kim CK. Cystic Fibrosis: A case presented with recurrent bronchiolitis in infancy in a Korean male infant. J Korean Med Sci. 1988; 3:157–62.
Article16. Ahn KM, Park HY, Lee JH, Lee MG, Kim JH, Kang IJ, et al. Cystic fibrosis in Korean children: A case report identified by a quantitative pilocarpine inotophoresis sweat test and genetic analysis. J Korean Med Sci. 2005; 20:153–7.17. Hwang IO, Lee ES. A case of cystic fibrosis presented with meconium ileus in a female neonate. Korean J Pediatr. 2007; 50:1252–6.
Article18. Ko JM, Kim GH, Kim KM, Hong SJ, Yoo HW. Identification of a novel mutation of CFTR gene in a Korean patient with cystic fibrosis. J Korean Med Sci. 2008; 23:912–5.19. Choe YJ, Ko JS, Seo JK, Han JJ, Shim JO, Koh YY, et al. Novel CFTR mutations in a Korean infant with cystic fibrosis and pancreatic insufficiency. J Korean Med Sci. 2010; 25:163–5.
Article20. Gee HY, Kim CK, Kim SW, Lee JH, Kim JH, Kim KH, et al. The L441P mutation of cystic fibrosis transmembrane conductance regulator and its molecular pathogenic mechanisms in a Korean patient with cystic fibrosis. J Korean Med Sci. 2010; 25:166–71.
Article21. Ranganathan SC, Stocks J, Dezateux C, Bush A, Wade A, Carr S, et al. The evolution of airway function in early childhood following clinical diagnosis of cystic fibrosis. Am J Respir Cirt Care Med. 2004; 169:928–33.22. Grosse SD, Rosenfeld M, Devine OJ, Lai HJ, Farrell PM. Potential impact of newborn screening for cystic fibrosis on child survival: A systematic review and analysis. J pediatr. 2006; 149:362–6.23. Grosse SD, Rosenfeld M, Devine OJ, Lai HJ, Farrell PM. Potential impact of newborn screening for cystic fibrosis on child survival: A systematic review and analysis. J pediatr. 2006; 149:362–6.24. Proesmans M, Vermeulen F, Boeck KD. What's new in cystic fibrosis? From treating symptoms to correction of the basic defects. Eur J Pediatr. 2008; 167:839–49.25. Mogayzel PJ Jr, Flume PA. Update in cystic fibrosis 2009. Am J Respir Crit Care Med. 2010; 181:593–44.
Article26. Zielenski J. Genotype and phenotype in cystic fibrosis. Respiration. 2000. 117–33.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Cystic fibrosis in a female adolescent carrying c.263T>G (p.Leu88X) and c.2977G>T (p.Asp993Tyr) mutation
- Report of a Korean Patient with Cystic Fibrosis, Carrying Q98R and Q220X Mutations in the CFTR Gene
- Identification of a Novel Mutation of CFTR Gene in a Korean Patient with Cystic Fibrosis
- F508 amino acid deletion mutation of CFTR gene in Korean lung cancer patients
- Expression of Cystic Fibrosis Transmembrane Regulator in Nasal Polyps
