Clinical Diversity of SCN4A-Mutation-Associated Skeletal Muscle Sodium Channelopathy
- Affiliations
-
- 1Department of Neurology, Dong-Eui Hospital, Busan, Korea.
- 2Department of Neurology, Medical Research Institute, Pusan National University School of Medicine, Busan, Korea. dskim@pusan.ac.kr
- 3Department of Neurology, College of Medicine, Yonsei University, Seoul, Korea.
- KMID: 2287648
- DOI: http://doi.org/10.3988/jcn.2009.5.4.186
Abstract
- BACKGROUND AND PURPOSE
Mutations of the skeletal muscle sodium channel gene SCN4A, which is located on chromosome 17q23-25, are associated with various neuromuscular disorders that are labeled collectively as skeletal muscle sodium channelopathy. These disorders include hyperkalemic periodic paralysis (HYPP), hypokalemic periodic paralysis, paramyotonia congenita (PMC), potassium-aggravated myotonia, and congenital myasthenic syndrome. This study analyzed the clinical and mutational spectra of skeletal muscle sodium channelopathy in Korean subjects. METHODS: Six unrelated Korean patients with periodic paralysis or nondystrophic myotonia associated with SCN4A mutations were included in the study. For the mutational analysis of SCN4A, we performed a full sequence analysis of the gene using the patients' DNA. We also analyzed the patients' clinical history, physical findings, laboratory tests, and responses to treatment. RESULTS: We identified four different mutations (one of which was novel) in all of the patients examined. The novel heterozygous missense mutation, p.R225W, was found in one patient with mild nonpainful myotonia. Our patients exhibited various clinical phenotypes: pure myotonia in four, and PMC in one, and HYPP in one. The four patients with pure myotonia were initially diagnosed as having myotonia congenita (MC), but a previous analysis revealed no CLCN1 mutation. CONCLUSIONS: Clinical differentiating between sodium-channel myotonia (SCM) and MC is not easy, and it is suggested that a mutational analysis of both SCN4A and CLCN1 is essential for the differential diagnosis of SCM and MC.
MeSH Terms
-
Channelopathies
Diagnosis, Differential
DNA
Humans
Hypokalemic Periodic Paralysis
Muscle, Skeletal
Mutation, Missense
Myasthenic Syndromes, Congenital
Myotonia
Myotonia Congenita
Myotonic Disorders
Paralyses, Familial Periodic
Paralysis
Paralysis, Hyperkalemic Periodic
Sequence Analysis
Sodium
Sodium Channels
DNA
Myotonia
Sodium
Sodium Channels
Figure
-
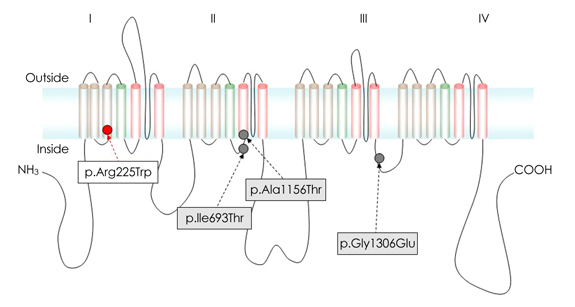
Fig. 1 Membrane-folding model of the sodium channel alpha subunit and locations of the missense mutations identified in the present study. A novel mutation, p.Arg 225Trp, is located at the transmembrane S3 segment of domain I.
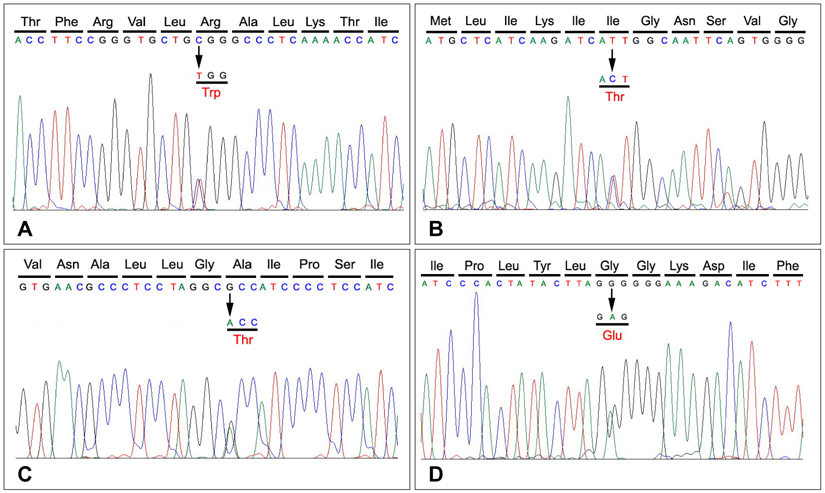
Fig. 2 Chromatograms of the patients. A: A novel mutation in exon 5 was identified in patient 3. A substitution of cytosine to thymidine at position 673 changes the codon for arginine at position 225 into tryptophan. B: A substitution of thymidine to cytosine at position 2078 in patient 4 changes the codon for isoleucine at position 693 into threonine. C: A substitution of guanine to adenine at position 3466 in patients 2 and 5 changes the codon for alanine at position 1156 into threonine. D: A substitution of guanine to adenine at position 3917 in patients 1 and 6 changes the codon for glycine at position 1306 into glutamate.

Fig. 3 A: The results of PCR-restriction fragment length polymorphism using restriction enzyme Aci I for the identification of the novel c.673C>T (p.Arg225Trp) mutation. No similar digestion pattern was observed among 100 normal control chromosomes. M, 100-bp molecular marker; U, undigested PCR product from patient 3; P, patient 3; numbers, control. B: Multiple alignment of the homologous sodium channel alpha subunit protein between different species of eukaryotes. The mutated protein in patient 3 (p.Arg 225Trp, arrowheads) is highly conserved between species.
Cited by 3 articles
-
The Overlap between Fibromyalgia Syndrome and Myotonia Congenita
Tai-Seung Nam, Seok-Yong Choi, Dong-Jin Park, Shin-Seok Lee, Young-Ok Kim, Myeong-Kyu Kim
J Clin Neurol. 2015;11(2):188-191. doi: 10.3988/jcn.2015.11.2.188.Whole-Body Muscle MRI in Patients with Hyperkalemic Periodic Paralysis Carrying the SCN4A Mutation T704M: Evidence for Chronic Progressive Myopathy with Selective Muscle Involvement
Young Han Lee, Hyung-Soo Lee, Hyo Eun Lee, Seok Hahn, Tai-Seung Nam, Ha Young Shin, Young-Chul Choi, Seung Min Kim
J Clin Neurol. 2015;11(4):331-338. doi: 10.3988/jcn.2015.11.4.331.Clinical Characteristics and Analysis of CLCN1 in Patients with "EMG Disease"
Tai-Seung Nam, Hyun-Jung Jung, Seok-Yong Choi, Young-Ok Kim, Myeong-Kyu Kim, Ki-Hyun Cho
J Clin Neurol. 2012;8(3):212-217. doi: 10.3988/jcn.2012.8.3.212.
Reference
-
1. George AL Jr, Ledbetter DH, Kallen RG, Barchi RL. Assignment of a human skeletal muscle sodium channel alpha-subunit gene (SCN4A) to 17q23.1-25.3. Genomics. 1991. 9:555–556.
Article2. George AL Jr, Crackower MA, Abdalla JA, Hudson AJ, Ebers GC. Molecular basis of Thomsen's disease (autosomal dominant myotonia congenita). Nat Genet. 1993. 3:305–310.
Article3. Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NS, et al. Human Gene Mutation Database (HGMD): 2003 update. Hum Mutat. 2003. 21:577–581.
Article4. Ptácek LJ, George AL Jr, Griggs RC, Tawil R, Kallen RG, Barchi RL, et al. Identification of a mutation in the gene causing hyperkalemic periodic paralysis. Cell. 1991. 67:1021–1027.
Article5. McClatchey AI, McKenna-Yasek D, Cros D, Worthen HG, Kuncl RW, DeSilva SM, et al. Novel mutations in families with unusual and variable disorders of the skeletal muscle sodium channel. Nat Genet. 1992. 2:148–152.
Article6. Orrell RW, Jurkat-Rott K, Lehmann-Horn F, Lane RJ. Familial cramp due to potassium-aggravated myotonia. J Neurol Neurosurg Psychiatry. 1998. 65:569–572.
Article7. Bulman DE, Scoggan KA, van Oene MD, Nicolle MW, Hahn AF, Tollar LL, et al. A novel sodium channel mutation in a family with hypokalemic periodic paralysis. Neurology. 1999. 53:1932–1936.
Article8. Tsujino A, Maertens C, Ohno K, Shen XM, Fukuda T, Harper CM, et al. Myasthenic syndrome caused by mutation of the SCN4A sodium channel. Proc Natl Acad Sci U S A. 2003. 100:7377–7382.9. Cannon SC. Spectrum of sodium channel disturbances in the nondystrophic myotonias and periodic paralyses. Kidney Int. 2000. 57:772–779.
Article10. Moon IS, Kim HS, Shin JH, Park YE, Park KH, Shin YB, et al. Novel CLCN1 mutations and clinical features of Korean patients with myotonia congenita. J Korean Med Sci. 2009. 24:1038–1044.
Article11. Wu FF, Takahashi MP, Pegoraro E, Angelini C, Colleselli P, Cannon SC, et al. A new mutation in a family with cold-aggravated myotonia disrupts Na(+) channel inactivation. Neurology. 2001. 56:878–884.
Article12. Rosenfeld J, Sloan-Brown K, George AL Jr. A novel muscle sodium channel mutation causes painful congenital myotonia. Ann Neurol. 1997. 42:811–814.
Article13. Ptacek LJ, Gouw L, Kwieciński H, McManis P, Mendell JR, Barohn RJ, et al. Sodium channel mutations in paramyotonia congenita and hyperkalemic periodic paralysis. Ann Neurol. 1993. 33:300–307.
Article14. Plassart E, Eymard B, Maurs L, Hauw JJ, Lyon-Caen O, Fardeau M, et al. Paramyotonia congenita: genotype to phenotype correlations in two families and report of a new mutation in the sodium channel gene. J Neurol Sci. 1996. 142:126–133.
Article15. McClatchey AI, McKenna-Yasek D, Cros D, Worthen HG, Kuncl RW, DeSilva SM, et al. Novel mutations in families with unusual and variable disorders of the skeletal muscle sodium channel. Nat Genet. 1992. 2:148–152.
Article16. Lerche H, Heine R, Pika U, George AL Jr, Mitrovic N, Browatzki M, et al. Human sodium channel myotonia: slowed channel inactivation due to substitutions for a glycine within the III-IV linker. J Physiol. 1993. 470:13–22.
Article17. Mitrović N, George AL Jr, Lerche H, Wagner S, Fahlke C, Lehmann-Horn F. Different effects on gating of three myotonia-causing mutations in the inactivation gate of the human muscle sodium channel. J Physiol. 1995. 487:107–114.
Article18. Rüdel R, Lehmann-Horn F. Paramyotonia, potassium-aggravated myotonias and periodic paralyses. 37th ENMC International Workshop, Naarden, The Netherlands, 8-10 December 1995. Neuromuscul Disord. 1997. 7:127–132.19. Schoser BG, Schröder JM, Grimm T, Sternberg D, Kress W. A large German kindred with cold-aggravated myotonia and a heterozygous A1481D mutation in the SCN4A gene. Muscle Nerve. 2007. 35:599–606.
Article20. Trip J, Drost G, Verbove DJ, van der Kooi AJ, Kuks JB, Notermans NC, et al. In tandem analysis of CLCN1 and SCN4A greatly enhances mutation detection in families with non-dystrophic myotonia. Eur J Hum Genet. 2008. 16:921–929.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Familial hyperkalemic periodic paralysis caused by a de novo mutation in the sodium channel gene SCN4A
- An atypical phenotype of hypokalemic periodic paralysis caused by a mutation in the sodium channel gene SCN4A
- A recurrent case of SCN4A related Paramyotonia congenita in two Korean brothers: a case report
- The Genotype and Clinical Phenotype of Korean Patients with Familial Hypokalemic Periodic Paralysis
- Unusual Voltage-Gated Sodium and Potassium Channelopathies Related to Epilepsy
