Compound Heterozygosity for Two Novel SLC26A4 Mutations in a Large Iranian Pedigree with Pendred Syndrome
- Affiliations
-
- 1Department of Biochemistry, Falavarjan Branch, Islamic Azad University, Isfahan, Iran.
- 2Cellular and Molecular Research Center, School of Medicine, Shahrekord University of Medical Sciences, Shahrekord, Iran. mchalesh@yahoo.com
- 3Department of Medical Genetics, School of Medicine, Ahvaz Jundishapur University of Medical Sciences, Ahvaz, Iran.
- 4Department of Biology, Science and Research Branch, Islamic Azad University, Tehran, Iran.
Abstract
OBJECTIVES
The aim of this study was to detect the genetic cause of deafness in a large Iranian family. Due to the importance of SLC26A4 in causing hearing loss, information about the gene mutations can be beneficial in molecular detection and management of deaf patients.
METHODS
We investigated the genetic etiology in a large consanguineous family with 9 deaf patients from Fars province of Iran with no GJB2 mutations. Initially, linkage analysis was performed by four DFNB4 short tandem repeat markers. The result showed linkage to DFNB4 locus. Following that, DNA sequencing of all 21 exons, their adjacent intronic sequences and the promoter of SLC26A4 was carried out for mutation detection.
RESULTS
Two novel mutations (c.863-864insT and c.881-882delAC) were identified in exon 7 of the gene, in both homozygous and compound heterozygous state in patients.
CONCLUSION
Our results supported the importance of the SLC26A4 mutations in the etiology of hearing loss among the Iranian patients and therefore its mutation screening should be considered after GJB2 in the molecular diagnostics of hearing loss, especially when enlarged vestibular aqueduct or goiter is detected.
Keyword
MeSH Terms
Figure
-
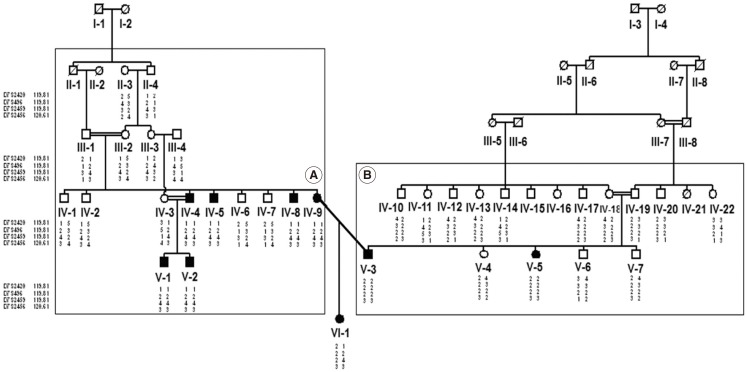
Fig. 1 Pedigree and haplotypes of the family Iranian 3 (IR3). The order of markers is based on the Marshfield map. The homozygous haplotype of part A was different from that of part B of the pedigree. Variants c.881-882delAC and c.863-864insT were found in part A and B, respectively. Patient VI-1 had both haplotypes simultaneously which were later shown to carry c.881-882delAC and c.863-864insT mutations (compound heterozygosity).
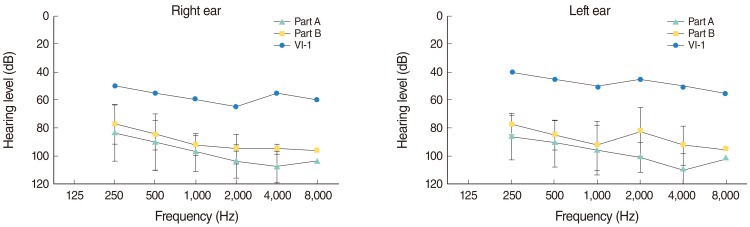
Fig. 2 Mean audiometric hearing thresholds for right and left ears of all patients of part A (triangles), patients V-3 and V-5 of part B (squares), and audiogram of both ears of patient VI-1 (circles) of the family Iranian 3.

Fig. 3 Thyroid ultrasonography result of patient V-5 (from family Iranian 3) with multinodular goiter. The arrows show nodules. The view is axial, and ultrasound probe is positioned in the neck region.

Fig. 4 Computed tomography scan of the temporal bone for patient IV-8 (from family Iranian 3) with bilateral enlarged vestibular aqueduct.

Fig. 5 Multipoint logarithm of odds (LOD) score calculation of the family Iranian 3 with SimWalk ver. 2.91 showed a score of 6.57 confirming linkage to DFNB4 locus.
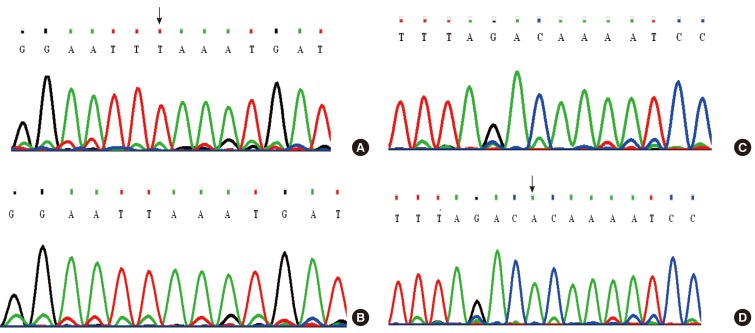
Fig. 6 Electropherogram results of two novel SLC26A4 variants in family Iranian 3 and normal alleles. (A) A patient with homozygous c.863-864insT allele. (B) A normal subject without c.863-864insT allele. (C) A patient with homozygous c.881-882delAC allele. (D) A normal subject without c.881-882delAC allele.
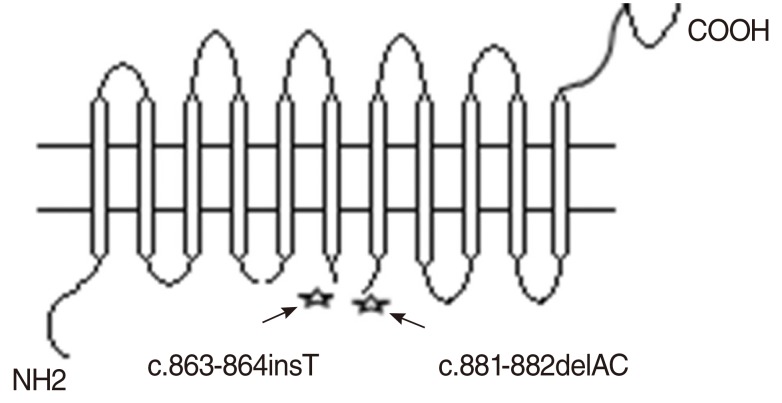
Fig. 7 c.863-864insT and c.881-882delAC variants which cause amino acid change at intracellular region (most probably, between the sixth and seventh transmembrane domains) of the pendrin protein in the Everett model.
Reference
-
1. Hilgert N, Smith RJ, Van Camp G. Forty-six genes causing nonsyndromic hearing impairment: which ones should be analyzed in DNA diagnostics? Mutat Res. 2009; Mar-Jun. 681(2-3):189–196. PMID: 18804553.
Article2. Bizhanova A, Kopp P. Genetics and phenomics of Pendred syndrome. Mol Cell Endocrinol. 2010; 6. 322(1-2):83–90. PMID: 20298745.
Article3. Everett LA, Glaser B, Beck JC, Idol JR, Buchs A, Heyman M, et al. Pendred syndrome is caused by mutations in a putative sulphate trans-porter gene (PDS). Nat Genet. 1997; 12. 17(4):411–422. PMID: 9398842.
Article4. Campbell C, Cucci RA, Prasad S, Green GE, Edeal JB, Galer CE, et al. Pendred syndrome, DFNB4, and PDS/SLC26A4 identification of eight novel mutations and possible genotype-phenotype correlations. Hum Mutat. 2001; 5. 17(5):403–411. PMID: 11317356.5. Phelps PD, Coffey RA, Trembath RC, Luxon LM, Grossman AB, Britton KE, et al. Radiological malformations of the ear in Pendred syndrome. Clin Radiol. 1998; 4. 53(4):268–273. PMID: 9585042.
Article6. Denoyelle F, Weil D, Maw MA, Wilcox SA, Lench NJ, Allen-Powell DR, et al. Prelingual deafness: high prevalence of a 30delG mutation in the connexin 26 gene. Hum Mol Genet. 1997; 11. 6(12):2173–2177. PMID: 9336442.
Article7. Frei K, Ramsebner R, Lucas T, Hamader G, Szuhai K, Weipoltshammer K, et al. GJB2 mutations in hearing impairment: identification of a broad clinical spectrum for improved genetic counseling. Laryngoscope. 2005; 3. 115(3):461–465. PMID: 15744158.
Article8. Marlin S, Garabédian EN, Roger G, Moatti L, Matha N, Lewin P, et al. Connexin 26 gene mutations in congenitally deaf children: pitfalls for genetic counseling. Arch Otolaryngol Head Neck Surg. 2001; 8. 127(8):927–933. PMID: 11493200.9. Hashemzadeh Chaleshtori M, Farhud DD, Patton MA. Familial and sporadic GJB2-related deafness in Iran: review of gene mutations. Iran J Public Health. 2007; 1. 36(1):1–14.10. Najmabadi H, Nishimura C, Kahrizi K, Riazalhosseini Y, Malekpour M, Daneshi A, et al. GJB2 mutations: passage through Iran. Am J Med Genet A. 2005; 3. 133A(2):132–137. PMID: 15666300.
Article11. Anwar S, Riazuddin S, Ahmed ZM, Tasneem S, Ateeq-ul-Jaleel , Khan SY, et al. SLC26A4 mutation spectrum associated with DFNB4 deaf-ness and Pendred's syndrome in Pakistanis. J Hum Genet. 2009; 5. 54(5):266–270. PMID: 19287372.
Article12. Park HJ, Shaukat S, Liu XZ, Hahn SH, Naz S, Ghosh M, et al. Origins and frequencies of SLC26A4 (PDS) mutations in east and south Asians: global implications for the epidemiology of deafness. J Med Genet. 2003; 4. 40(4):242–248. PMID: 12676893.
Article13. Kahrizi K, Mohseni M, Nishimura C, Bazazzadegan N, Fischer SM, Dehghani A, et al. Identification of SLC26A4 gene mutations in Iranian families with hereditary hearing impairment. Eur J Pediatr. 2009; 6. 168(6):651–653. PMID: 18813951.
Article14. Park HJ, Lee SJ, Jin HS, Lee JO, Go SH, Jang HS, et al. Genetic basis of hearing loss associated with enlarged vestibular aqueducts in Koreans. Clin Genet. 2005; 2. 67(2):160–165. PMID: 15679828.
Article15. Wang QJ, Zhao YL, Rao SQ, Guo YF, Yuan H, Zong L, et al. A distinct spectrum of SLC26A4 mutations in patients with enlarged vestibular aqueduct in China. Clin Genet. 2007; 9. 72(3):245–254. PMID: 17718863.
Article16. Berrettini S, Forli F, Bogazzi F, Neri E, Salvatori L, Casani AP, et al. Large vestibular aqueduct syndrome: audiological, radiological, clinical, and genetic features. Am J Otolaryngol. 2005; Nov-Dec. 26(6):363–371. PMID: 16275403.
Article17. Grimberg J, Nawoschik S, Belluscio L, McKee R, Turck A, Eisenberg A. A simple and efficient non-organic procedure for the isolation of genomic DNA from blood. Nucleic Acids Res. 1989; 10. 17(20):8390. PMID: 2813076.
Article18. Lindner TH, Hoffmann K. easyLINKAGE: a PERL script for easy and automated two-/multi-point linkage analyses. Bioinformatics. 2005; 2. 21(3):405–407. PMID: 15347576.
Article19. Fishelson M, Geiger D. Optimizing exact genetic linkage computations. J Comput Biol. 2004; 3. 11(2-3):263–275. PMID: 15285892.
Article20. Thiele H, Nurnberg P. HaploPainter: a tool for drawing pedigrees with complex haplotypes. Bioinformatics. 2005; 4. 21(8):1730–1732. PMID: 15377505.
Article21. Scott DA, Wang R, Kreman TM, Andrews M, McDonald JM, Bishop JR, et al. Functional differences of the PDS gene product are associated with phenotypic variation in patients with Pendred syndrome and non-syndromic hearing loss (DFNB4). Hum Mol Genet. 2000; 7. 9(11):1709–1715. PMID: 10861298.
Article22. Pera A, Dossena S, Rodighiero S, Gandia M, Botta G, Meyer G, et al. Functional assessment of allelic variants in the SLC26A4 gene involved in Pendred syndrome and nonsyndromic EVA. Proc Natl Acad Sci U S A. 2008; 11. 105(47):18608–18613. PMID: 19017801.
Article23. Taylor JP, Metcalfe RA, Watson PF, Weetman AP, Trembath RC. Mutations of the PDS gene, encoding pendrin, are associated with protein mislocalization and loss of iodide efflux: implications for thyroid dysfunction in Pendred syndrome. J Clin Endocrinol Metab. 2002; 4. 87(4):1778–1784. PMID: 11932316.24. Hutchin T, Coy NN, Conlon H, Telford E, Bromelow K, Blaydon D, et al. Assessment of the genetic causes of recessive childhood non-syndromic deafness in the UK: implications for genetic testing. Clin Genet. 2005; 12. 68(6):506–512. PMID: 16283880.25. Alasti F, Peeters N, Wuyts W, Sanati MH, Van Camp G. Novel human pathological mutations. Gene symbol: SLC26A4. Disease: deafness, non-syndromic, autosomal recessive. Hum Genet. 2010; 1. 127(1):116. PMID: 20108392.26. Tabatabaiefar MA, Alasti F, Peeters N, Wuyts W, Nooridaloii MR, Chaleshtori MH, et al. Novel human pathological mutations. Gene symbol: SLC26A4. Disease: Pendred syndrome. Hum Genet. 2010; 4. 127(4):468–469. PMID: 21488234.
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Genetic Screening of GJB2 and SLC26A4 in Korean Cochlear Implantees: Experience of Soree Ear Clinic
- SLC26A4 Mutations in Korean Population
- Carrier frequency of SLC26A4 mutations causing inherited deafness in the Korean population
- A Family of H723R Mutation for SLC26A4 Associated with Enlarged Vestibular Aqueduct Syndrome
- Novel Compound Heterozygous Mutations in CTSC Gene in a Chinese Family with Papillon–Lefevre Syndrome
