AnsNGS: An Annotation System to Sequence Variations of Next Generation Sequencing Data for Disease-Related Phenotypes
- Affiliations
-
- 1Seoul National University Biomedical Informatics (SNUBI), Division of Biomedical Informatics, Seoul National University College of Medicine, Seoul, Korea. juhan@snu.ac.kr
- 2Systems Biomedical Informatics Research Center, Seoul National University, Seoul, Korea.
- KMID: 2166663
- DOI: http://doi.org/10.4258/hir.2013.19.1.50
Abstract
OBJECTIVES
Next-generation sequencing (NGS) data in the identification of disease-causing genes provides a promising opportunity in the diagnosis of disease. Beyond the previous efforts for NGS data alignment, variant detection, and visualization, developing a comprehensive annotation system supported by multiple layers of disease phenotype-related databases is essential for deciphering the human genome. To satisfy the impending need to decipher the human genome, it is essential to develop a comprehensive annotation system supported by multiple layers of disease phenotype-related databases.
METHODS
AnsNGS (Annotation system of sequence variations for next-generation sequencing data) is a tool for contextualizing variants related to diseases and examining their functional consequences. The AnsNGS integrates a variety of annotation databases to attain multiple levels of annotation.
RESULTS
The AnsNGS assigns biological functions to variants, and provides gene (or disease)-centric queries for finding disease-causing variants. The AnsNGS also connects those genes harbouring variants and the corresponding expression probes for downstream analysis using expression microarrays. Here, we demonstrate its ability to identify disease-related variants in the human genome.
CONCLUSIONS
The AnsNGS can give a key insight into which of these variants is already known to be involved in a disease-related phenotype or located in or near a known regulatory site. The AnsNGS is available free of charge to academic users and can be obtained from http://snubi.org/software/AnsNGS/.
Keyword
MeSH Terms
Figure
-
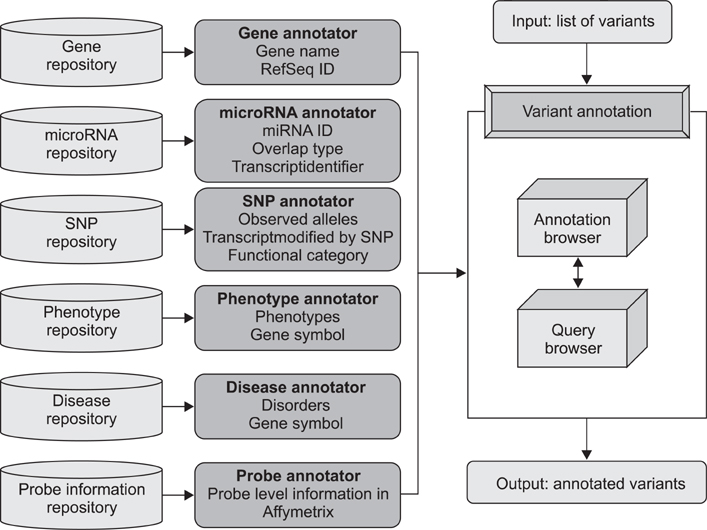
Figure 1 Overview of the AnsNGS. The AnsNGS takes text-based input files, obtains annotation information from the integrated databases, and returns the detailed annotation output to the user. The AnsNGS queries various tables in the UCSC database to extract the information for the reference genome or any data set conforming to Generic Feature Format ver. 3 (GFF3). The system consists of six annotators. The six annotators in the AnsNGS are a gene annotator, a microRNA annotator, an SNP annotator, a phenotype annotator, a disease annotator, and a probe annotator. NGS: next-generation sequencing, SNP: single nucleotide polymorphism.
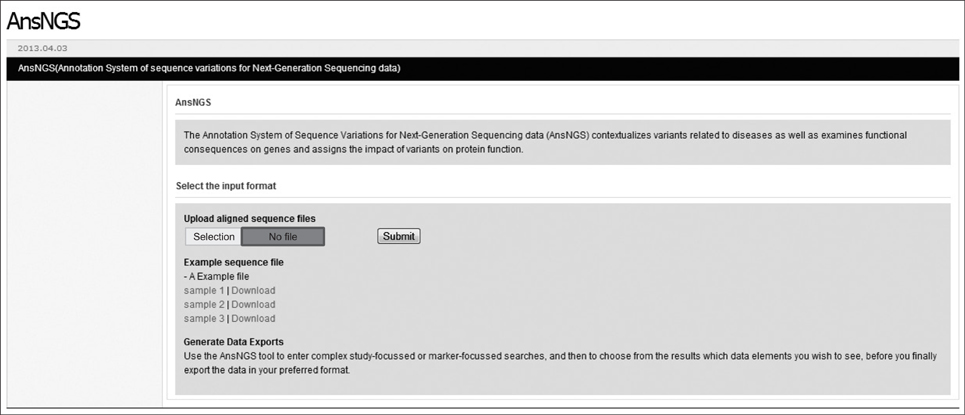
Figure 2 Interface of the AnsNGS. The main page of the AnsNGS is composed of three examples of sequencing data, such as whole genome or whole exome sequencing data. The example files are downloadable. The input file format of the AnsNGS is described in the Methods section. NGS: next-generation sequencing.
Reference
-
1. Kaiser J. DNA sequencing: a plan to capture human diversity in 1000 genomes. Science. 2008. 319(5862):395.2. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010. 38(16):e164.
Article3. Shetty AC, Athri P, Mondal K, Horner VL, Steinberg KM, Patel V, et al. SeqAnt: a Web service to rapidly identify and annotate DNA sequence variations. BMC Bioinformatics. 2010. 11:471.
Article4. Sana ME, Iascone M, Marchetti D, Palatini J, Galasso M, Volinia S. GAMES identifies and annotates mutations in next-generation sequencing projects. Bioinformatics. 2011. 27(1):9–13.
Article5. Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, et al. The human genome browser at UCSC. Genome Res. 2002. 12(6):996–1006.
Article6. Griffiths-Jones S. miRBase: the microRNA sequence database. Methods Mol Biol. 2006. 342:129–138.
Article7. Becker KG, Barnes KC, Bright TJ, Wang SA. The genetic association database. Nat Genet. 2004. 36(5):431–432.
Article8. Hamosh A, Scott AF, Amberger J, Valle D, McKusick VA. Online Mendelian Inheritance in Man (OMIM). Hum Mutat. 2000. 15(1):57–61.
Article9. Okou DT, Steinberg KM, Middle C, Cutler DJ, Albert TJ, Zwick ME. Microarray-based genomic selection for high-throughput resequencing. Nat Methods. 2007. 4(11):907–909.
Article10. Ng SB, Turner EH, Robertson PD, Flygare SD, Bigham AW, Lee C, et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009. 461(7261):272–276.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- SFannotation: A Simple and Fast Protein Function Annotation System
- A Primer for Disease Gene Prioritization Using Next-Generation Sequencing Data
- A Simple GUI-based Sequencing Format Conversion Tool for the Three NGS Platforms
- CaGe: A Web-Based Cancer Gene Annotation System for Cancer Genomics
- Molecular genetic decoding of malformations of cortical development
