Exome Sequencing Reveals a Novel PRPS1 Mutation in a Family with CMTX5 without Optic Atrophy
- Affiliations
-
- 1Department of Neurology, Ewha Womans University School of Medicine, Seoul, Korea.
- 2Department of Biological Science, Kongju National University, Gongju, Korea. kwchung@kongju.ac.kr
- 3Department of Neurology, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea. bochoi@skku.edu
- KMID: 1980550
- DOI: http://doi.org/10.3988/jcn.2013.9.4.283
Abstract
- BACKGROUND
X-linked Charcot-Marie-Tooth disease type 5 (CMTX5) is caused by mutations in the gene encoding phosphoribosyl pyrophosphate synthetase I (PRPS1). There has been only one case report of CMTX5 patients. The aim of this study was to identify the causative gene in a family with CMTX with peripheral neuropathy and deafness.
CASE REPORT
A Korean family with X-linked recessive CMT was enrolled. The age at the onset of hearing loss of the male proband was 5 months, and that of steppage gait was 6 years; he underwent cochlear surgery at the age of 12 years. In contrast to what was reported for the first patients with CMTX5, this patient did not exhibit optic atrophy. Furthermore, there was no cognitive impairment, respiratory dysfunction, or visual disturbance. Assessment of his family history revealed two male relatives with very similar clinical manifestations. Electrophysiological evaluations disclosed sensorineural hearing loss and peripheral neuropathy. Whole-exome sequencing identified a novel p.Ala121Gly (c.362C>G) PRPS1 mutation as the underlying genetic cause of the clinical phenotype.
CONCLUSIONS
A novel mutation of PRPS1 was identified in a CMTX5 family in which the proband had a phenotype of peripheral neuropathy with early-onset hearing loss, but no optic atrophy. The findings of this study will expand the clinical spectrum of X-linked recessive CMT and will be useful for the molecular diagnosis of clinically heterogeneous peripheral neuropathies.
Keyword
MeSH Terms
Figure
-
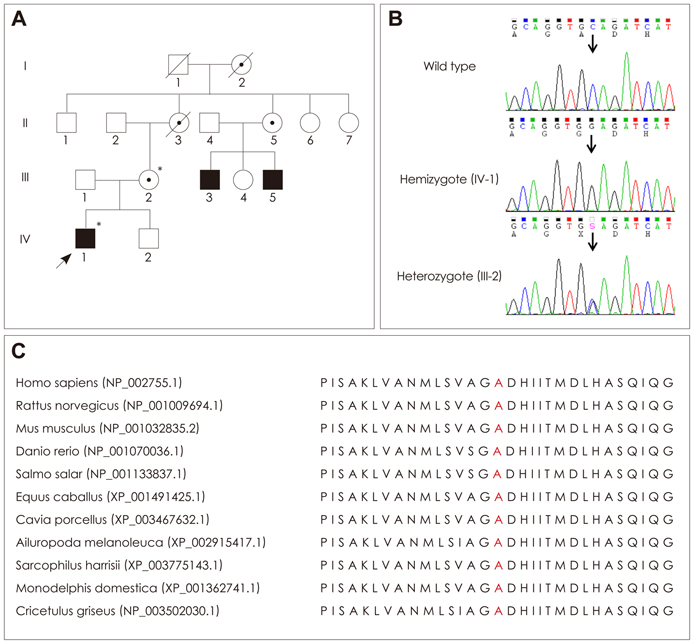
Fig. 1 Pedigree, sequencing, and conservation analysis. A: Pedigree of a Korean family with X-linked Charcot-Marie-Tooth disease type 5. Open symbols, unaffected individuals; filled symbols, affected individuals; circles with a dot inside, putative carriers of PRPS1 mutation; arrow, proband; asterisks, individuals whose DNA was used for DNA sequencing. B: Sequencing chromatograms showing the PRPS1c.362C>G (Ala-121Gly) mutation. The affected proband (IV-1) is hemizygous, whereas his unaffected mother is heterozygous for this locus. Vertical arrows indicate the mutation site. C: Conservation of amino acid sequences among different species. The analysis was performed using MEGA5 (ver. 5.05) software. The mutation site and its neighboring sequences were highly conserved among the different species.
Reference
-
1. Kim HJ, Sohn KM, Shy ME, Krajewski KM, Hwang M, Park JH, et al. Mutations in PRPS1, which encodes the phosphoribosyl pyrophosphate synthetase enzyme critical for nucleotide biosynthesis, cause hereditary peripheral neuropathy with hearing loss and optic neuropathy (cmtx5). Am J Hum Genet. 2007; 81:552–558.
Article2. Rosenberg RN, Chutorian A. Familial opticoacoustic nerve degeneration and polyneuropathy. Neurology. 1967; 17:827–832.
Article3. Sperling O, Eilam G, Sara-Persky-Brosh , De Vries A. Accelerated erythrocyte 5-phosphoribosyl-1-pyrophosphate synthesis. A familial abnormality associated with excessive uric acid production and gout. Biochem Med. 1972; 6:310–316.
Article4. Arts WF, Loonen MC, Sengers RC, Slooff JL. X-linked ataxia, weakness, deafness, and loss of vision in early childhood with a fatal course. Ann Neurol. 1993; 33:535–539.
Article5. de Brouwer AP, Williams KL, Duley JA, van Kuilenburg AB, Nabuurs SB, Egmont-Petersen M, et al. Arts syndrome is caused by loss-of-function mutations in PRPS1. Am J Hum Genet. 2007; 81:507–518.
Article6. Liu X, Han D, Li J, Han B, Ouyang X, Cheng J, et al. Loss-of-function mutations in the PRPS1 gene cause a type of nonsyndromic X-linked sensorineural deafness, DFN2. Am J Hum Genet. 2010; 86:65–71.
Article7. de Brouwer AP, van Bokhoven H, Nabuurs SB, Arts WF, Christodoulou J, Duley J. PRPS1 mutations: four distinct syndromes and potential treatment. Am J Hum Genet. 2010; 86:506–518.
Article8. Becker MA, Heidler SA, Bell GI, Seino S, Le Beau MM, Westbrook CA, et al. Cloning of cDNAs for human phosphoribosylpyrophosphate synthetases 1 and 2 and X chromosome localization of PRPS1 and PRPS2 genes. Genomics. 1990; 8:555–561.
Article9. Lee JH, Choi BO. Charcot-marie-tooth disease: seventeen causative genes. J Clin Neurol. 2006; 2:92–106.
Article10. Montenegro G, Powell E, Huang J, Speziani F, Edwards YJ, Beecham G, et al. Exome sequencing allows for rapid gene identification in a Charcot-Marie-Tooth family. Ann Neurol. 2011; 69:464–470.
Article11. Choi BO, Koo SK, Park MH, Rhee H, Yang SJ, Choi KG, et al. Exome sequencing is an efficient tool for genetic screening of Charcot-Marie-Tooth disease. Hum Mutat. 2012; 33:1610–1615.
Article12. Becker MA, Puig JG, Mateos FA, Jimenez ML, Kim M, Simmonds HA. Inherited superactivity of phosphoribosylpyrophosphate synthetase: association of uric acid overproduction and sensorineural deafness. Am J Med. 1988; 85:383–390.
Article13. Kim HJ, Hong SH, Ki CS, Kim BJ, Shim JS, Cho SH, et al. A novel locus for X-linked recessive CMT with deafness and optic neuropathy maps to Xq21.32-q24. Neurology. 2005; 64:1964–1967.
Article14. Duley JA, Christodoulou J, de Brouwer AP. The PRPP synthetase spectrum: what does it demonstrate about nucleotide syndromes? Nucleosides Nucleotides Nucleic Acids. 2011; 30:1129–1139.
Article15. Bottiglieri T. S-Adenosyl-L-methionine (SAMe): from the bench to the bedside--molecular basis of a pleiotrophic molecule. Am J Clin Nutr. 2002; 76:1151S–1157S.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Three Cases of Leber's Hereditary Optic Atrophy in One Family
- Diagnostic Odyssey and Application of Targeted Exome Sequencing in the Investigation of Recurrent Infant Deaths in a Syrian Consanguineous Family: a Case of Spinal Muscular Atrophy with Respiratory Distress Type 1
- A Mitochondrial Mutation in Leber's Hereditary Optic Neuropathy
- Exome Sequencing in Mendelian Disorders
- Whole Exome Sequencing of a Patient with Duchenne Muscular Dystrophy
