Tuberc Respir Dis.
2012 Sep;73(3):162-168. 10.4046/trd.2012.73.3.162.
Clinical Predictors of Survival in Idiopathic Pulmonary Fibrosis
- Affiliations
-
- 1Department of Internal Medicine, Ewha Womans University School of Medicine, Seoul, Korea. jinhwalee@ewha.ac.kr
- KMID: 1842911
- DOI: http://doi.org/10.4046/trd.2012.73.3.162
Abstract
- BACKGROUND
Idiopathic pulmonary fibrosis (IPF) is a progressive disease. Effective treatment is not currently available and the prognosis is poor. The aim of our study was to identify clinical predictors of survival in patients with IPF.
METHODS
By using medical record database of a university hospital, we reviewed the records of patients who had been diagnosed as having IPF from January 1996 through December 2007.
RESULTS
Among 89 patients considered as having interstitial lung disease (ILD) on computed tomography (CT) of the chest, 22 were excluded because of the diagnosis of other ILDs or connective tissue disease, and finally, 67 met the criteria of IPF. The mean age at the diagnosis of IPF was 70 years (range, 41~87 years) and 43 (64%) were male. The mean survival time following the diagnosis of IPF was 40 months (range, 0~179 months). Among them, 28 cases were diagnosed as the progressive state of IPF on the follow-up CT examination, and the mean duration between diagnosis of IPF and progression was 31 months. Multivariate analysis using Cox regression model revealed that body mass index (BMI) less than 18.5 kg/m2 (p=0.030; hazard ratio [HR], 12.085; 95% confidence interval [CI], 1.277~114.331) and CT progression before 36 months from the diagnosis of IPF (p=0.042; HR, 13.564; 95% CI, 1.101~167.166) were independently associated with mortality.
CONCLUSION
Since low BMI at the diagnosis of IPF and progression on follow-up CT were associated with poor prognosis, IPF patients with low BMI and/or progression before 36 months following the diagnosis should be closely monitored.
MeSH Terms
Figure
-
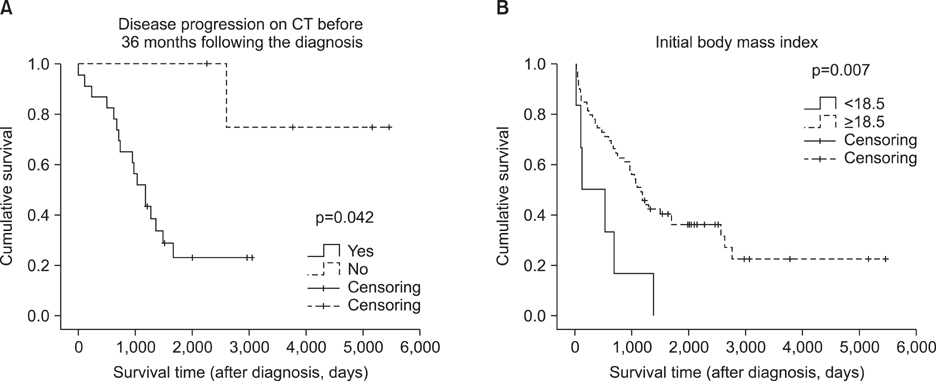
Figure 1 Comparison of overall survival by Kaplan-Meier method. (A) Disease progression on computed tomography (CT) before 36 months following the diagnosis. (B) Initial body mass index. p-value by Log rank test.
Reference
-
1. American Thoracic Society. European Respiratory Society. American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am J Respir Crit Care Med. 2002. 165:277–304.2. Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011. 183:788–824.3. American Thoracic Society. Standardization of Spirometry, 1994 Update. Am J Respir Crit Care Med. 1995. 152:1107–1136.4. Kim DS, Collard HR, King TE Jr. Classification and natural history of the idiopathic interstitial pneumonias. Proc Am Thorac Soc. 2006. 3:285–292.5. Nagai S, Nagao T, Kitaichi M, Izumi T. Clinical courses of asymptomatic cases with idiopathic pulmonary fibrosis and a histology of usual interstitial pneumonia. Eur Respir J. 1998. 11:131s.6. Bjoraker JA, Ryu JH, Edwin MK, Myers JL, Tazelaar HD, Schroeder DR, et al. Prognostic significance of histopathologic subsets in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1998. 157:199–203.7. Nagai S, Kitaichi M, Hamada K, Nagao T, Hoshino Y, Miki H, et al. Hospital-based historical cohort study of 234 histologically proven Japanese patients with IPF. Sarcoidosis Vasc Diffuse Lung Dis. 1999. 16:209–214.8. Collard HR, King TE Jr, Bartelson BB, Vourlekis JS, Schwarz MI, Brown KK. Changes in clinical and physiologic variables predict survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2003. 168:538–542.9. Flaherty KR, Mumford JA, Murray S, Kazerooni EA, Gross BH, Colby TV, et al. Prognostic implications of physiologic and radiographic changes in idiopathic interstitial pneumonia. Am J Respir Crit Care Med. 2003. 168:543–548.10. Jegal Y, Kim DS, Shim TS, Lim CM, Lee SD, Koh Y, et al. Physiology is a stronger predictor of survival than pathology in fibrotic interstitial pneumonia. Am J Respir Crit Care Med. 2005. 171:639–644.11. Nicholson AG, Colby TV, Dubois RM, Hansell DM, Wells AU. The prognostic significance of the histologic pattern of interstitial pneumonia in patients presenting with the clinical entity of cryptogenic fibrosing alveolitis. Am J Respir Crit Care Med. 2000. 162:2213–2217.12. Travis WD, Matsui K, Moss J, Ferrans VJ. Idiopathic nonspecific interstitial pneumonia: prognostic significance of cellular and fibrosing patterns: survival comparison with usual interstitial pneumonia and desquamative interstitial pneumonia. Am J Surg Pathol. 2000. 24:19–33.13. Raghu G, Brown KK, Bradford WZ, Starko K, Noble PW, Schwartz DA, et al. A placebo-controlled trial of interferon gamma-1b in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2004. 350:125–133.14. Demedts M, Behr J, Buhl R, Costabel U, Dekhuijzen R, Jansen HM, et al. High-dose acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med. 2005. 353:2229–2242.15. Scientific Committee of the Korean Academy of Tuberculosis and Respiratory Diseases. 2008 National Survey of Idiopathic Interstitial Pneumonia in Korea. Tuberc Respir Dis. 2009. 66:141–151.16. Panos RJ, Mortenson RL, Niccoli SA, King TE Jr. Clinical deterioration in patients with idiopathic pulmonary fibrosis: causes and assessment. Am J Med. 1990. 88:396–404.17. Kondoh Y, Taniguchi H, Kawabata Y, Yokoi T, Suzuki K, Takagi K. Acute exacerbation in idiopathic pulmonary fibrosis. Analysis of clinical and pathologic findings in three cases. Chest. 1993. 103:1808–1812.18. Ambrosini V, Cancellieri A, Chilosi M, Zompatori M, Trisolini R, Saragoni L, et al. Acute exacerbation of idiopathic pulmonary fibrosis: report of a series. Eur Respir J. 2003. 22:821–826.19. Akira M, Hamada H, Sakatani M, Kobayashi C, Nishioka M, Yamamoto S. CT findings during phase of accelerated deterioration in patients with idiopathic pulmonary fibrosis. AJR Am J Roentgenol. 1997. 168:79–83.20. Azuma A, Nukiwa T, Tsuboi E, Suga M, Abe S, Nakata K, et al. Double-blind, placebo-controlled trial of pirfenidone in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2005. 171:1040–1047.21. Kim DS, Park JH, Park BK, Lee JS, Nicholson AG, Colby T. Acute exacerbation of idiopathic pulmonary fibrosis: frequency and clinical features. Eur Respir J. 2006. 27:143–150.22. Parambil JG, Myers JL, Ryu JH. Histopathologic features and outcome of patients with acute exacerbation of idiopathic pulmonary fibrosis undergoing surgical lung biopsy. Chest. 2005. 128:3310–3315.23. Kubo H, Nakayama K, Yanai M, Suzuki T, Yamaya M, Watanabe M, et al. Anticoagulant therapy for idiopathic pulmonary fibrosis. Chest. 2005. 128:1475–1482.24. Kondoh Y, Taniguchi H, Yokoi T, Nishiyama O, Ohishi T, Kato T, et al. Cyclophosphamide and low-dose prednisolone in idiopathic pulmonary fibrosis and fibrosing nonspecific interstitial pneumonia. Eur Respir J. 2005. 25:528–533.25. Martinez FJ, Safrin S, Weycker D, Starko KM, Bradford WZ, King TE Jr, et al. The clinical course of patients with idiopathic pulmonary fibrosis. Ann Intern Med. 2005. 142(12 Pt 1):963–967.26. King TE Jr, Schwarz MI, Brown K, Tooze JA, Colby TV, Waldron JA Jr, et al. Idiopathic pulmonary fibrosis: relationship between histopathologic features and mortality. Am J Respir Crit Care Med. 2001. 164:1025–1032.27. Flaherty KR, Colby TV, Travis WD, Toews GB, Mumford J, Murray S, et al. Fibroblastic foci in usual interstitial pneumonia: idiopathic versus collagen vascular disease. Am J Respir Crit Care Med. 2003. 167:1410–1415.28. Nicholson AG, Fulford LG, Colby TV, du Bois RM, Hansell DM, Wells AU. The relationship between individual histologic features and disease progression in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2002. 166:173–177.29. World Health Organization. Obesity: preventing and managing the global epidemic. Report on a WHO Consultation. WHO Technical Report Series No. 894. 2000. Geneva: World Health Organization.30. Nici L, Donner C, Wouters E, Zuwallack R, Ambrosino N, Bourbeau J, et al. American Thoracic Society/European Respiratory Society statement on pulmonary rehabilitation. Am J Respir Crit Care Med. 2006. 173:1390–1413.31. Stark LJ, Powers SW. Behavioral aspects of nutrition in children with cystic fibrosis. Curr Opin Pulm Med. 2005. 11:539–542.32. Davis PB. Cystic fibrosis since 1938. Am J Respir Crit Care Med. 2006. 173:475–482.33. Cano NJ, Pichard C, Roth H, Court-Fortuné I, Cynober L, Gérard-Boncompain M, et al. C-reactive protein and body mass index predict outcome in end-stage respiratory failure. Chest. 2004. 126:540–546.34. Alakhras M, Decker PA, Nadrous HF, Collazo-Clavell M, Ryu JH. Body mass index and mortality in patients with idiopathic pulmonary fibrosis. Chest. 2007. 131:1448–1453.35. Tilg H, Moschen AR. Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat Rev Immunol. 2006. 6:772–783.36. Savino W. The thymus gland is a target in malnutrition. Eur J Clin Nutr. 2002. 56:Suppl 3. S46–S49.37. Arcasoy SM, Christie JD, Ferrari VA, Sutton MS, Zisman DA, Blumenthal NP, et al. Echocardiographic assessment of pulmonary hypertension in patients with advanced lung disease. Am J Respir Crit Care Med. 2003. 167:735–740.38. Mejía M, Carrillo G, Rojas-Serrano J, Estrada A, Suárez T, Alonso D, et al. Idiopathic pulmonary fibrosis and emphysema: decreased survival associated with severe pulmonary arterial hypertension. Chest. 2009. 136:10–15.39. Cottin V, Nunes H, Brillet PY, Delaval P, Devouassoux G, Tillie-Leblond I, et al. Combined pulmonary fibrosis and emphysema: a distinct underrecognised entity. Eur Respir J. 2005. 26:586–593.
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Two cases of idiopathic pulmonary fibrosis
- Idiopathic Pulmonary Fibrosis and Nonspecific Interstitial Pneumonia
- An ROC study detecting ability of idiopathic pulmonary fibrosis using digital radiography
- Pharmacological treatment of idiopathic pulmonary fibrosis and fibrosing interstitial lung diseases: current trends and future directions
- Diagnosis and treatment of interstitial lung disease: focusing on idiopathic pulmonary fibrosis
