Ann Lab Med.
2014 Sep;34(5):390-394. 10.3343/alm.2014.34.5.390.
A de novo Microdeletion of ANKRD11 Gene in a Korean Patient with KBG Syndrome
- Affiliations
-
- 1Medical Genetics Center, University of Ulsan College of Medicine and Asan Medical Center, Seoul, Korea. ejseo@amc.seoul.kr
- 2Department of Laboratory Medicine, University of Ulsan College of Medicine and Asan Medical Center, Seoul, Korea.
- 3Department of Pediatrics, University of Ulsan College of Medicine and Asan Medical Center, Seoul, Korea.
- 4Department of Laboratory Medicine, Ulsan University Hospital, University of Ulsan College of Medicine, Ulsan, Korea.
- 5Asan Institute for Life Sciences, Asan Medical Center, Seoul, Korea.
- 6Department of Pediatrics, Pusan National University Children's Hospital, Busan, Korea.
- KMID: 1791956
- DOI: http://doi.org/10.3343/alm.2014.34.5.390
Abstract
- KBG syndrome is a very rare genetic disorder characterized by macrodontia of upper central incisors, global developmental delay, distinctive craniofacial features, short stature, and skeletal anomalies. Ankyrin repeat domain 11 gene (ANKRD11) has recently been identified as a causal factor of this syndrome. We describe a 6-yr-old Korean boy with features of KBG syndrome. The patient had a short stature, macrodontia, dysmorphic facial features, speech and motor delay with intellectual disability, and partial seizures as indicated by the electroencephalogram, but he was neither autistic nor had autism spectrum disorders. Using high-resolution oligonucleotide array comparative genomic hybridization, we identified a heterozygous 240-kb deletion at 16q24.3 corresponding to ANKRD11. This patient provided additional evidence on the influence of ANKRD11 in KBG syndrome and suggested that deletion limited to ANKRD11 is unlikely to cause autism.
Keyword
MeSH Terms
-
Abnormalities, Multiple/diagnosis/*genetics
Asian Continental Ancestry Group/*genetics
Bone Diseases, Developmental/diagnosis/*genetics
Child
Chromosomes, Human, Pair 16
Comparative Genomic Hybridization
Electroencephalography
Facies
Gene Deletion
Heterozygote
Humans
Intellectual Disability/diagnosis/*genetics
Male
Phenotype
Repressor Proteins/*genetics
Republic of Korea
Tooth Abnormalities/diagnosis/*genetics
Repressor Proteins
Figure
-
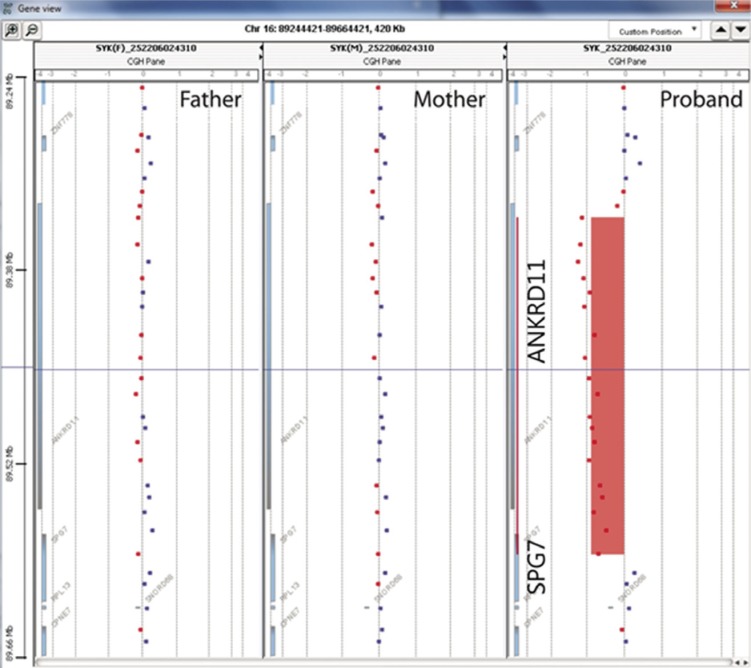
Fig. 1 Array comparative genomic hybridization data showing a 240-kb deletion in the proband. The red bar is shifted to a log2 ratio of -1 in the proband and indicates the deletion overlapping ANKRD11 and SPG7 at 16q24.3.The results of both parents indicate normal copy number.
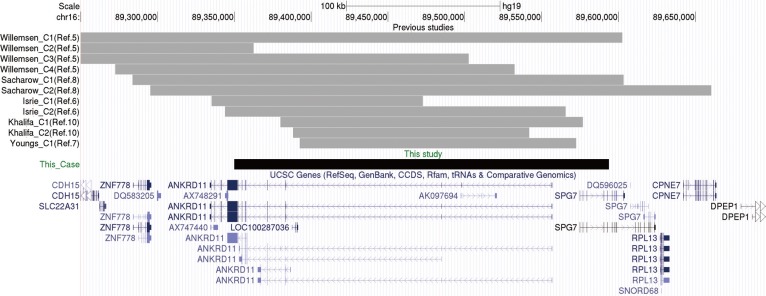
Fig. 2 Deletion boundaries in the patient reported in this study (black box covers exons 2-13 of ANKRD11 and exons 1-5 of SPG7) and previously reported cases (gray boxes). The boxes indicate the deletion size. The map is based on genome build GRCH37/hg19.
Reference
-
1. Brancati F, D'Avanzo MG, Digilio MC, Sarkozy A, Biondi M, De Brasi D, et al. KBG syndrome in a cohort of Italian patients. Am J Med Genet A. 2004; 131:144–149.
Article2. Herrmann J, Pallister PD, Tiddy W, Opitz JM. The KBG syndrome-a syndrome of short stature, characteristic facies, mental retardation, macrodontia and skeletal anomalies. Birth Defects Orig Artic Ser. 1975; 11:7–18.3. Zollino M, Battaglia A, D'Avanzo MG, Della Bruna MM, Marini R, Scarano G, et al. Six additional cases of the KBG syndrome: clinical reports and outline of the diagnostic criteria. Am J Med Genet. 1994; 52:302–307.
Article4. Sirmaci A, Spiliopoulos M, Brancati F, Powell E, Duman D, Abrams A, et al. Mutations in ANKRD11 cause KBG syndrome, characterized by intellectual disability, skeletal malformations, and macrodontia. Am J Hum Genet. 2011; 89:289–294. PMID: 21782149.
Article5. Willemsen MH, Fernandez BA, Bacino CA, Gerkes E, de Brouwer AP, Pfundt R, et al. Identification of ANKRD11 and ZNF778 as candidate genes for autism and variable cognitive impairment in the novel 16q24.3 microdeletion syndrome. Eur J Hum Genet. 2010; 18:429–435.6. Isrie M, Hendriks Y, Gielissen N, Sistermans EA, Willemsen MH, Peeters H, et al. Haploinsufficiency of ANKRD11 causes mild cognitive impairment, short stature and minor dysmorphisms. Eur J Hum Genet. 2012; 20:131–133. PMID: 21654729.7. Youngs EL, Hellings JA, Butler MG. ANKRD11 gene deletion in a 17-year-old male. Clin Dysmorphol. 2011; 20:170–171.8. Sacharow S, Li D, Fan YS, Tekin M. Familial 16q24.3 microdeletion involving ANKRD11 causes a KBG-like syndrome. Am J med Genet A. 2012; 158A:547–552.9. Khalifa M, Stein J, Grau L, Nelson V, Meck J, Aradhya S, et al. Partial deletion of ANKRD11 results in the KBG phenotype distinct from the 16q24.3 microdeletion syndrome. Am J Med Genet A. 2013; 161A:835–840.10. Brancati F, Sarkozy A, Dallapiccola B. KBG syndrome. Orphanet J Rare Dis. 2006; 1:50.
Article11. Lo-Castro A, Brancati F, Digilio MC, Garaci FG, Bollero P, Alfieri P, et al. Neurobehavioral phenotype observed in KBG syndrome caused by ANKRD11 mutations. Am J Med Genet B Neuropsychiatr Genet. 2013; 162B:17–23.
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Early-Onset Parkinson’s Disease in a Patient With a De Novo Frameshift Variant of the ANKRD11 Gene and KBG Syndrome
- An ANKRD11 exonic deletion accompanied by a congenital megacolon in an infant with KBG syndrome
- Early Diagnosis of KBG Syndrome Using Diagnostic Exome Sequencing
- The first Korean case of 2p15p16.1 microdeletion syndrome, characterized by facial dysmorphism, developmental delay, and congenital hypothyroidism
- Identification of a Novel De Novo Mutation of the TAZ Gene in a Korean Patient with Barth Syndrome
