Type VII Collagen Gene Mutations (c.8569G>T and c.4879G>A) Result in the Moderately Severe Phenotype of Recessive Dystrophic Epidermolysis Bullosa in a Korean Patient
- Affiliations
-
- 1Department of Dermatology, Keimyung University, School of Medicine, Daegu, Korea. franzes@dsmc.or.kr
- 2Department of Dermatology, Hirosaki University School of Medicine, Hirosaki, Japan.
- KMID: 1779127
- DOI: http://doi.org/10.3346/jkms.2009.24.2.256
Abstract
- Dystrophic epidermolysis bullosa (DEB) are caused by mutations in the COL7A1 gene, which encodes type VII collagen. Even though more than 500 different COL7A1 mutations have been identified in DEB, it still remains to be under-investigated. To investigate the mutation of COL7A1 in moderately severe phenotype of recessive DEB (RDEB) in a Korean patient, the mutation detection strategy was consisted of polymerase chain reaction (PCR) amplification of genomic DNA, followed by heteroduplex analysis, nucleotide sequencing of the PCR products demonstrating altered mobility. In this study, we found that one mutation (c.8569G>T) was detected within exon 116. The mutation of c.8569G>T in exon 116 changed the GAG (Glu) to TAG, eventually resulted in premature termination of type VII collagen polypeptide. Furthermore the mother did not have the mutation c.8569G>T in exon 116. The other novel mutation (c.4879G>A) was detected within exon 51 of both patient and mother, thereby resulting in changing valine (Val) to isoleucine (Ile) in type VII collagen polypeptide. Taken together, in this study we identified compound heterozygosity for COL7A1 mutations (c.8569G>T and c.4879G>A) in moderately severe RDEB in a Korean patient. We hope that this data contribute to the expanding database on COL7A1 mutations in DEB.
Keyword
MeSH Terms
Figure
-
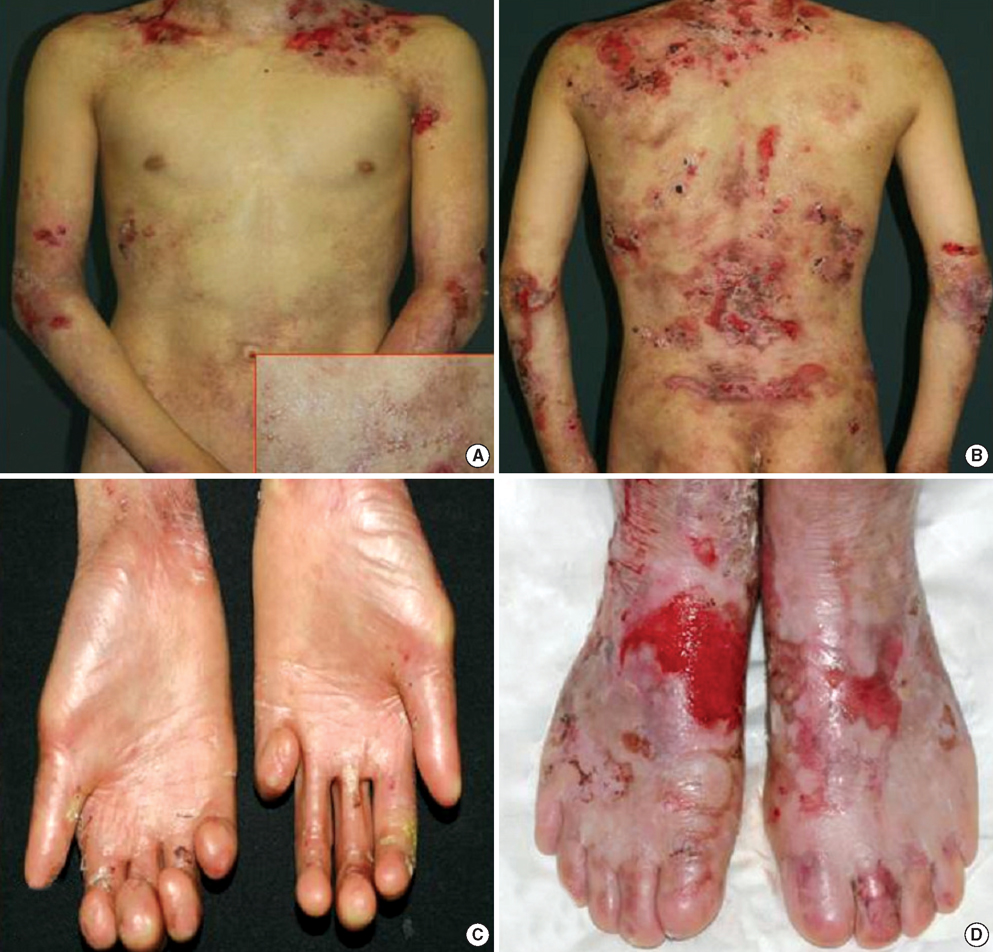
Fig. 1 Clinical features. Blisters and erosions were seen on the trunk (A, B) as well as milia (inset). Acral scaring and loss of finger and toe nail (C, D).
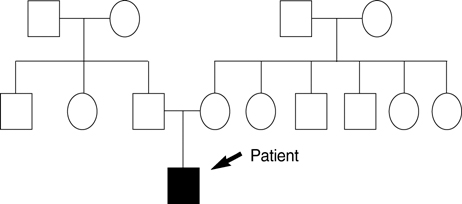
Fig. 2 The schematic presentation of the pedigree of the patient. The black arrow indicates the patient.
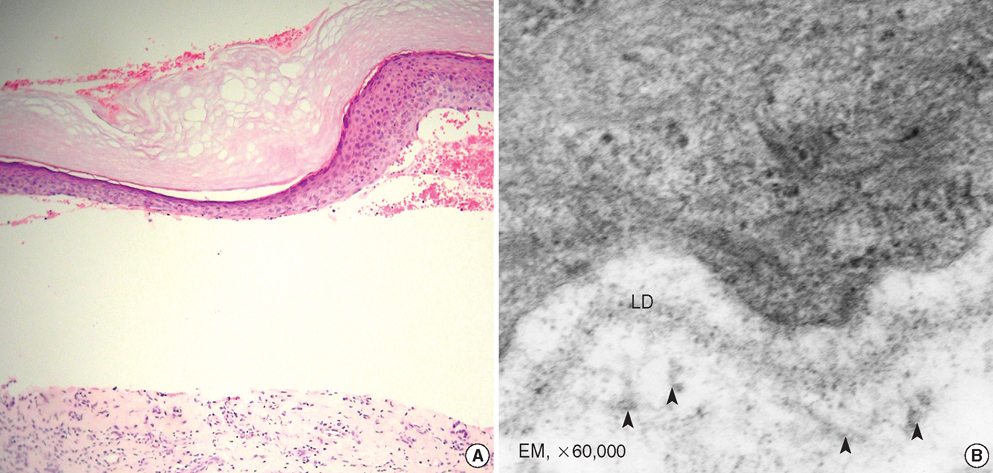
Fig. 3 Histological findings. Subepidermal blister formation and flattened rete ridges are observed (A) (H&E, ×200). Electron micrograph showing a few amount of anchoring fibrils (arrow heads) beneath the lamina densa (LD) (B).
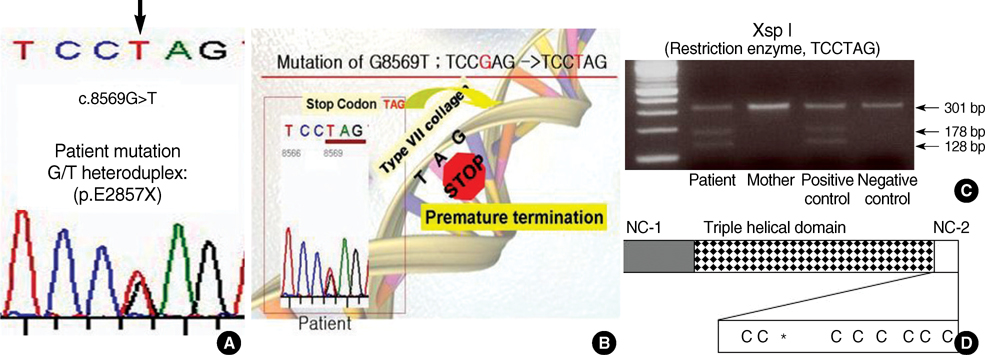
Fig. 4 Identification of the mutations in COL7A1 in patient. Direct sequencing of the patient PCR product revealed G to T transition at position 8569 (A). As a result, the codon for Glu (GAG) is changed to a codon for stop (TAG). This mutation was confirmed by restriction enzyme, XspI (C). Schematic representation of the domain structure of procollagen VII and the location of the premature stop codon (E2857X) in exon 116 (D). Cysteine residues are marked as a "C", and an asterisk (*) shows the procollagen C-proteinase cleavage site.
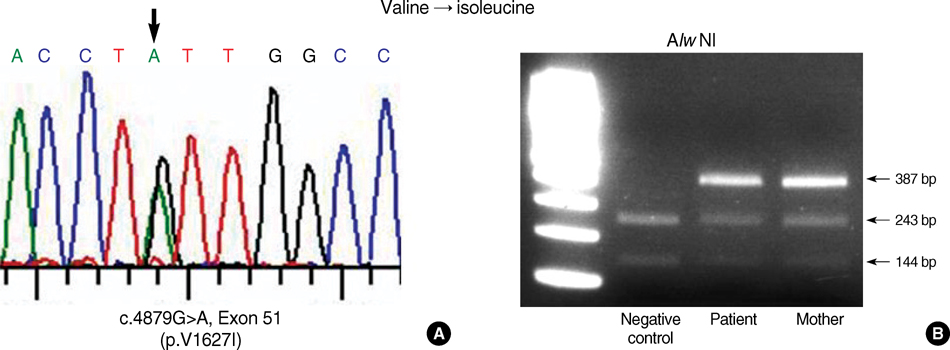
Fig. 5 Identification of the mutations in COL7A1 in the patient and mother. Direct sequencing of the patient PCR product revealed G to A transition at position 4879 (A). As a result, the codon for Val (GUU) is changed to a codon for Ile (AUU). This mutation was confirmed by restriction enzyme, Alw NI (B).
Reference
-
1. Uitto J, Pulkkinen L, Christiano AM. Molecular basis of the dystrophic and junctional forms of epidermolysis bullosa: mutations in the type VII collagen and kalinin (laminin 5) genes. J Invest Dermatol. 1994. 103:5 Suppl. 39S–46S.
Article2. Varki R, Sadowski S, Uitto J, Pfendner E. Epidermolysis bullosa. II. Type VII collagen mutations and phenotype-genotype correlations in the dystrophic subtypes. J Med Genet. 2007. 44:181–192.
Article3. Hamada T, Fukuda S, Ishii N, Sakaguchi S, Ishikawa T, Abe T, Yasumoto S, Hashimoto T, Nakano H, Sawamura D. Genotype-phenotype correlation in non-Hallopeau-Siemens recessive dystrophic epidermolysis bullosa: the splice site mutation c.6216+5G>T in the COL7A1 gene results in aberrant and normal splicings. J Dermatol Sci. 2008. 50:147–150.4. Uitto J, Hovnanian A, Christiano AM. Premature termination codon mutations in the type VII collagen gene (COL7A1) underlie severe recessive dystrophic epidermolysis bullosa. Proc Assoc Am Physicians. 1995. 107:245–252.5. Christiano AM, Amano S, Eichenfield LF, Burgeson RE, Uitto J. Premature termination codon mutations in the type VII collagen gene in recessive dystrophic epidermolysis bullosa result in nonsense-mediated mRNA decay and absence of functional protein. J Invest Dermatol. 1997. 109:390–394.
Article6. Hovnanian A, Rochat A, Bodemer C, Petit E, Rivers CA, Prost C, Fraitag S, Christiano AM, Uitto J, Lathrop M, Barrandon Y, de Prost Y. Characterization of 18 new mutations in COL7A1 in recessive dystrophic epidermolysis bullosa provides evidence for distinct molecular mechanisms underlying defective anchoring fibril formation. Am J Hum Genet. 1997. 61:599–610.7. Oh SW, Lee JS, Kim MY, Kim SC. COL7A1 mutational analysis in Korean patients with dystrophic epidermolysis bullosa. Br J Dermatol. 2007. 157:1260–1264.8. Christiano AM, McGrath JA, Tan KC, Uitto J. Glycine substitutions in the triple-helical region of type VII collagen result in a spectrum of dystrophic epidermolysis bullosa phenotypes and patterns of inheritance. Am J Hum Genet. 1996. 58:671–681.9. Terracina M, Posteraro P, Schubert M, Sonego G, Atzori F, Zambruno G, Bruckner-Tuderman L, Castiglia D. Compound heterozygosity for a recessive glycine substitution and a splice site mutation in the COL7A1 gene causes an unusually mild form of localized recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 1998. 111:744–750.10. Shimizu H, Hammami-Hauasli N, Hatta N, Nishikawa T, Bruckner-Tuderman L. Compound heterozygosity for silent and dominant glycine substitution mutations in COL7A1 leads to a marked transient intracytoplasmic retention of procollagen VII and a moderately severe dystrophic epidermolysis bullosa phenotype. J Invest Dermatol. 1999. 113:419–421.11. Kim J, Kim SC, Yasukawa K, Shimizu H. Compound heterozygosity for premature termination codon and glycine substitution mutations in the COL7A1 gene in Korean siblings with a moderately severe phenotype of recessive dystrophic epidermolysis bullosa. J Dermatol Sci. 2003. 33:180–183.12. Masunaga T, Shimizu H, Takizawa Y, Uitto J, Nishikawa T. Combination of novel premature termination codon and glycine substitution mutations in COL7A1 leads to moderately severe recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 2000. 114:204–205.13. Christiano AM, Hoffman GG, Zhang X, Xu Y, Tamai Y, Greenspan DS, Uitto J. Strategy for identification of sequence variants in COL7A1 and a novel 2-bp deletion mutation in recessive dystrophic epidermolysis bullosa. Hum Mutat. 1997. 10:408–414.14. Ganguly A, Rock MJ, Prockop DJ. Conformation-sensitive gel electrophoresis for rapid detection of single-base differences in double-stranded PCR products and DNA fragments: evidence for solvent-induced bends in DNA heteroduplexes. Proc Natl Acad Sci USA. 1993. 90:10325–10329.
Article15. Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA. 1977. 74:5463–5467.
Article16. Fine JD, McGrath J, Eady RA. Inherited epidermolysis bullosa comes into the new millenium: a revised classification system based on current knowledge of pathogenetic mechanisms and the clinical, laboratory, and epidemiologic findings of large, well-defined patient cohorts. J Am Acad Dermatol. 2000. 43:135–137.
Article17. Christiano AM, Greenspan DS, Lee S, Uitto J. Cloning of human type VII collagen. Complete primary sequence of the alpha 1(VII) chain and identification of intragenic polymorphisms. J Biol Chem. 1994. 269:20256–20262.
Article18. Christiano AM, Rosenbaum LM, Chung-Honet LC, Parente MG, Woodley DT, Pan TC, Zhang RZ, Chu ML, Burgeson RE, Uitto J. The large non-collagenous domain (NC-1) of type VII collagen is amino-terminal and chimeric. Homology to cartilage matrix protein, the type III domains of fibronectin and the A domains of von Willebrand factor. Hum Mol Genet. 1992. 1:475–481.
Article19. Hammami-Hauasli N, Schumann H, Raghunath M, Kilgus O, Luthi U, Luger T, Bruckner-Tuderman L. Some, but not all, glycine substitution mutations in COL7A1 result in intracellular accumulation of collagen VII, loss of anchoring fibrils, and skin blistering. J Biol Chem. 1998. 273:19228–19234.20. Burgeson RE. Type VII collagen, anchoring fibrils, and epidermolysis bullosa. J Invest Dermatol. 1993. 101:252–255.
Article21. Christiano AM, McGrath JA, Uitto J. Influence of the second COL7A1 mutation in determining the phenotypic severity of recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 1996. 106:766–770.22. Matsunaga A, Sasaki J, Han H, Huang W, Kugi M, Koga T, Ichiki S, Shinkawa T, Arakawa K. Compound heterozygosity for an apolipoprotein A1 gene promoter mutation and a structural nonsense mutation with apolipoprotein A1 deficiency. Arterioscler Thromb Vasc Biol. 1999. 19:348–355.
Article23. Tamai K, Ishida-Yamamoto A, Matsuo S, Iizuka H, Hashimoto I, Christiano AM, Uitto J, McGrath JA. Compound heterozygosity for a nonsense mutation and a splice site mutation in the type VII collagen gene (COL7A1) in recessive dystrophic epidermolysis bullosa. Lab Invest. 1997. 76:209–217.
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Transient Bullous Dermolysis of the Newborn
- Newborn with Dominant Dystrophic Epidermolysis Bullosa That Manifested as Aplasia Cutis Congenita at Birth
- Dystrophic Epidermolysis Bullosa in Two Sisters
- Missense Variant c.3301C>T (p.R1101W) in von Willebrand Factor A Sequence in a Patient with Recessive Dystrophic Epidermolysis Bullosa Pruriginosa with Compound Heterozygous COL7A1 Variants
- Pretibial Epidermolysis Bullosa with Nail Dystrophy in a Family
