Deficient Gap Junction Coupling in Two Common Hearing Loss-Related Variants of GJB2
- Affiliations
-
- 1Department of Otorhinolaryngology, The First Affiliated Hospital and Institute of Otorhinolaryngology, Sun Yat-sen University, Guangzhou, China
- 2Department of Otorhinolaryngology Head and Neck Surgery, Hainan General Hospital (Hainan Affiliated Hospital of Hainan Medical University), Haikou, China
- KMID: 2558836
- DOI: http://doi.org/10.21053/ceo.2023.00078
Abstract
Objectives
. The aim of this study was to explore the functional consequences of two common variants, p.V37I and c.299-300delAT, in the hearing loss-associated gene GJB2.
Methods
. Connexin 26 expression and gap junctional permeability were studied in HEK 293T cells transfected with plasmids expressing GJB2 wild-type, p.V37I, or c.299-300delAT CX26 proteins tagged with fluorescent markers. Functional analyses of various GJB2 haplotypes were conducted to thoroughly evaluate alterations in ionic and small-molecule coupling.
Results
. The p.V37I protein was localized at the plasma membrane, but it failed to effectively transport intercellular propidium iodide or Ca2+ efficiently, indicating an impairment in both biochemical and ionic coupling. The presence of GJB2 p.V37I seemed to increase the cells’ sensitivity to H2O2 treatment. In contrast, the known variant c.299-300delAT protein was not transported to the cell membrane and was unable to form gap junctions, remaining confined to the cytoplasm. Both ionic and biochemical coupling were defective in cells transfected with c.299-300delAT.
Conclusion
. The p.V37I and c.299-300delAT GJB2 mutations resulted in deficient gap junction-mediated coupling. Additionally, environmental factors could influence the functional outcomes of the GJB2 p.V37I mutation. These findings could pave the way for the development of molecular therapies targeting GJB2 mutations to treat hearing loss.
Figure
-
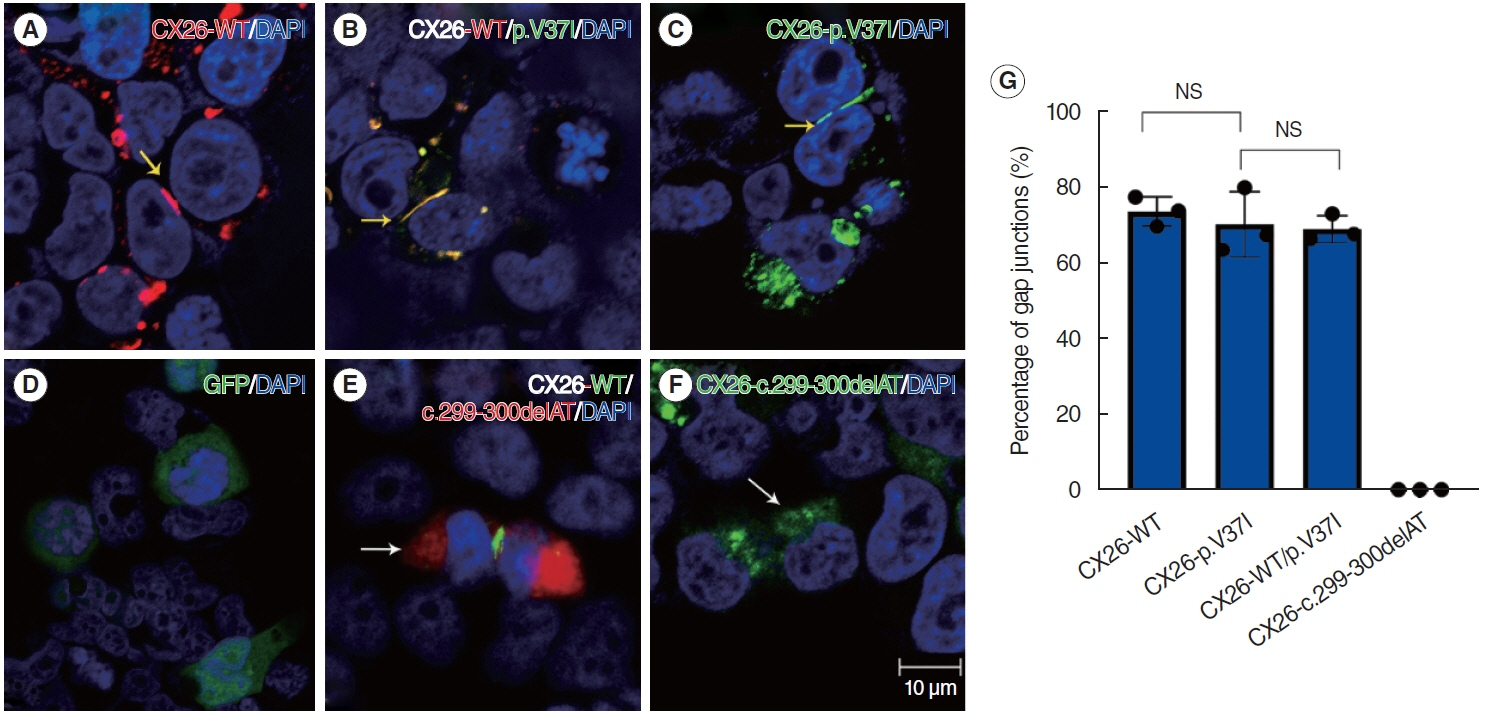
Fig. 1. (A-F) Immunofluorescence staining results after transfection with various CX26 vectors. Compared with the massive plaques in HEK 293T cells expressing CX26-c.299-300delAT (white arrows in E and F) or empty vector (D), intercellular gap junctions (yellow arrows in A-C) were observed. Scale bar=10 μm. (G) The percentages of gap junctions formed in three haplotype are shown. DAPI, 4´,6-diamidino-2´-phenylindole; GFP, green fluorescent protein; NS, not significant.
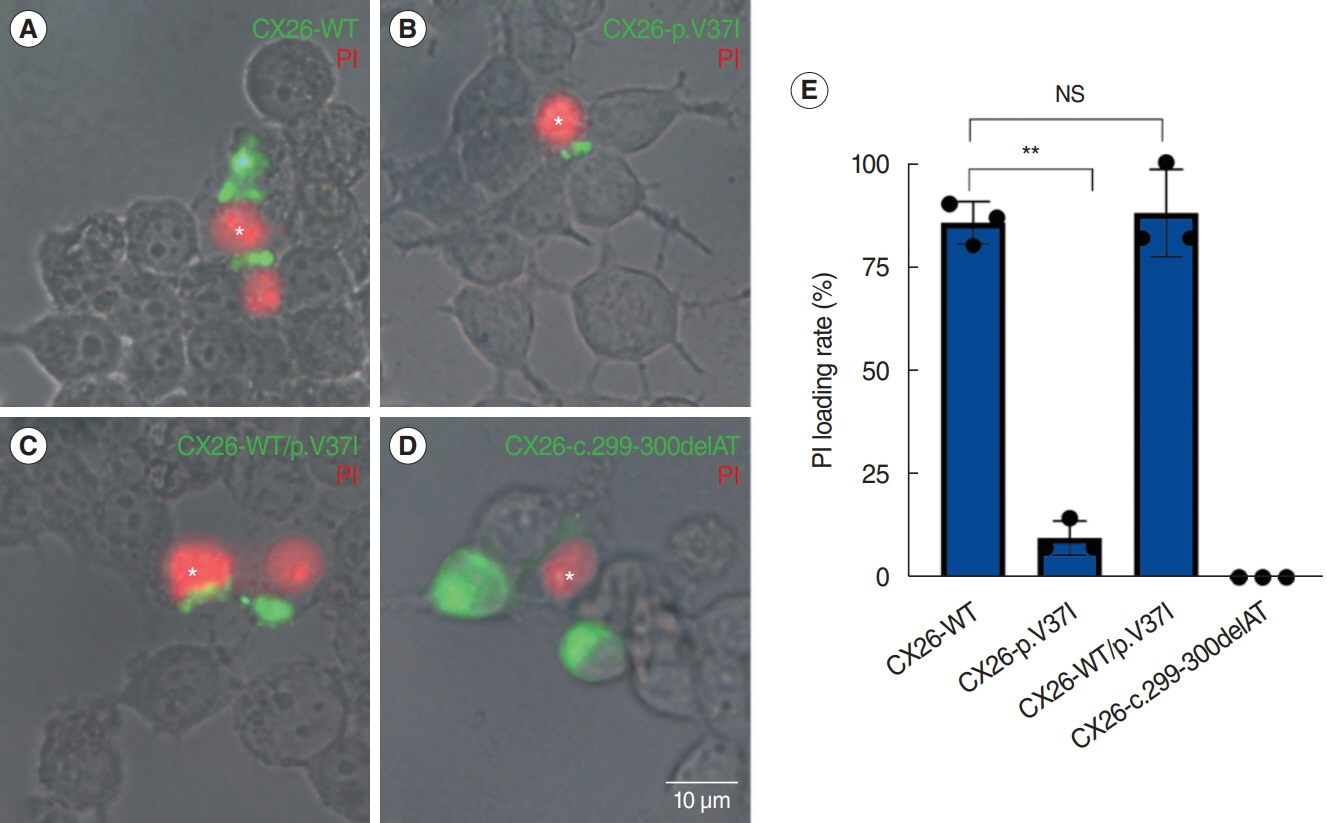
Fig. 2. Propidium iodide (PI) permeability by gap junctions formed by the four CX26 haplotypes in HEK 293T cells. Asterisks indicate cells that were injected with PI. (A-D) Representative images of cells expressing CX26-WT (A) or CX26-WT/p.V37I (C), showing the detection of PI dye in neighboring cells with gap junctions (green), in contrast to leaky PI transfer capacity in cells expressing CX26-p.V37I (B) or c.299-300delAT (D). Scale bar=10 μm. (E) Cell count analysis supporting the severe alteration in PI loading in CX26-p.V37I cells. NS, not significant. **P<0.01.
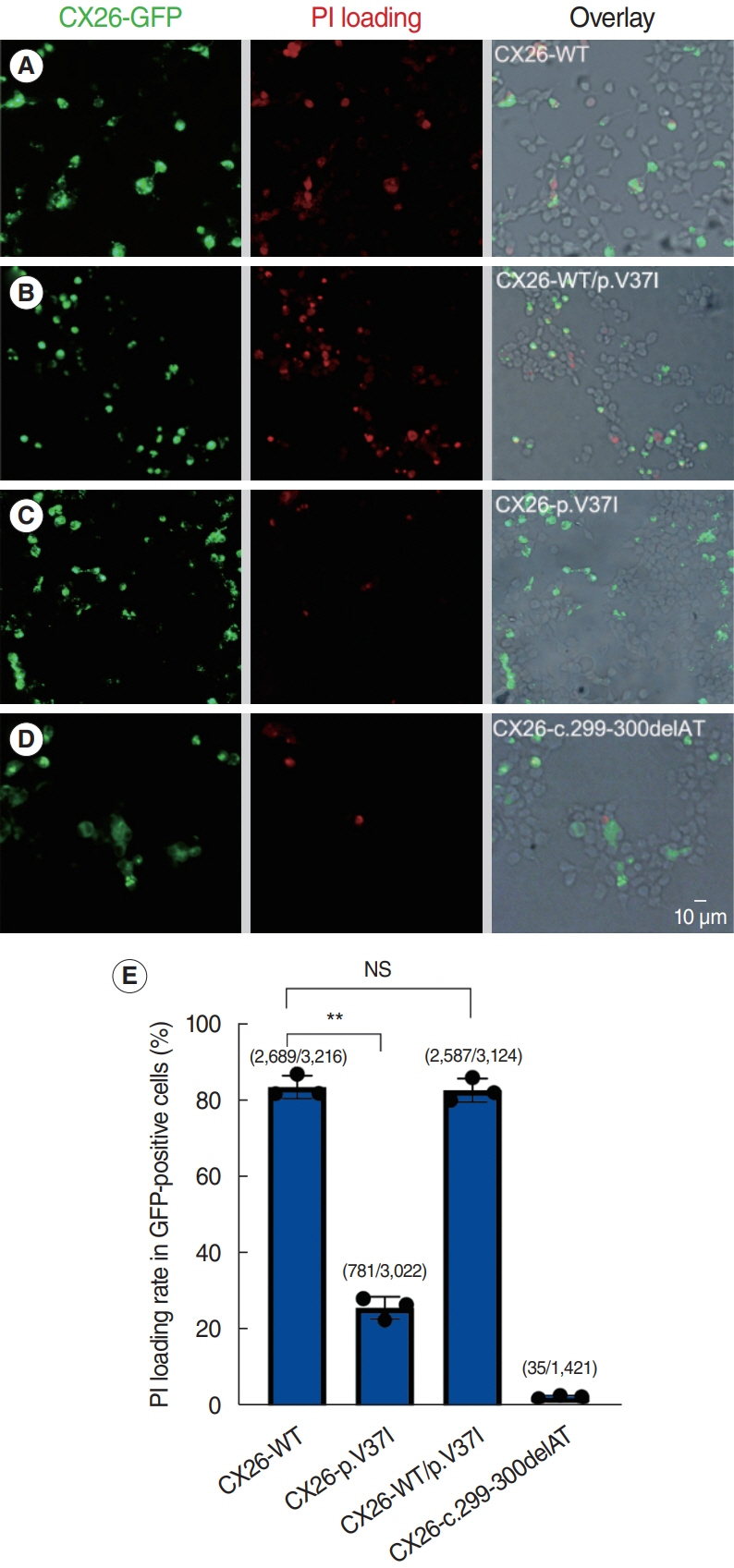
Fig. 3. Hemichannel dye loading assay of HEK 293T cells expressing the four connexin 26 (CX26) haplotypes. (A-D) Representative images of cells loaded with the membrane-impermeable fluorescent dye propidium iodide (PI). Scale bar=10 μm. (E) Quantification of the PI loading rate, showing a significant reduction in cells expressing CX26-p.V37I compared with that in cells expressing CX26-WT. NS, not significant. **P<0.01.
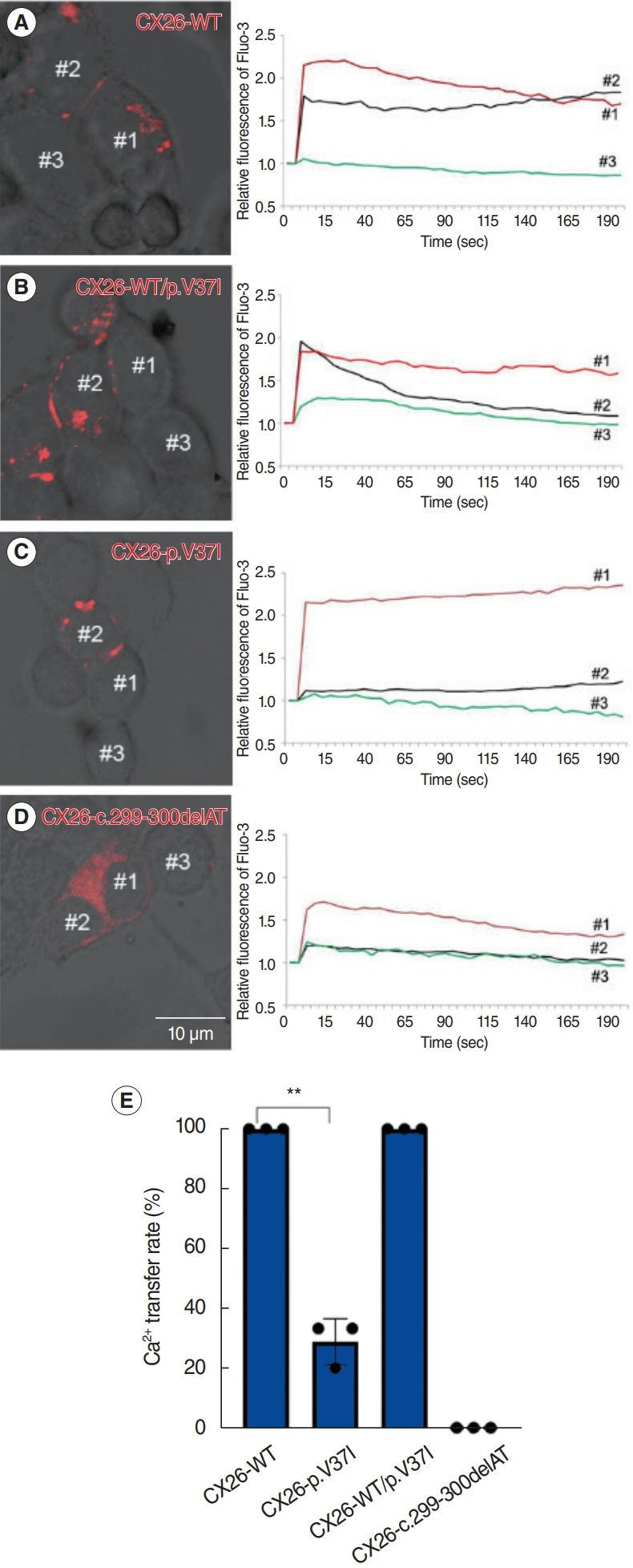
Fig. 4. (A, B) Ionic permeability of gap junctions (GJs) in HEK 293T cells were functional in cells expressing the CX26-WT and CX26-WT/p.V37I haplotypes. (C, D) Impaired permeability of ionic GJs was detected in CX26-p.V37I and CX26-c.299-300delAT. Scale bar= 10 μm. (E) The Ca2+ transfer rates are shown. **P<0.01.
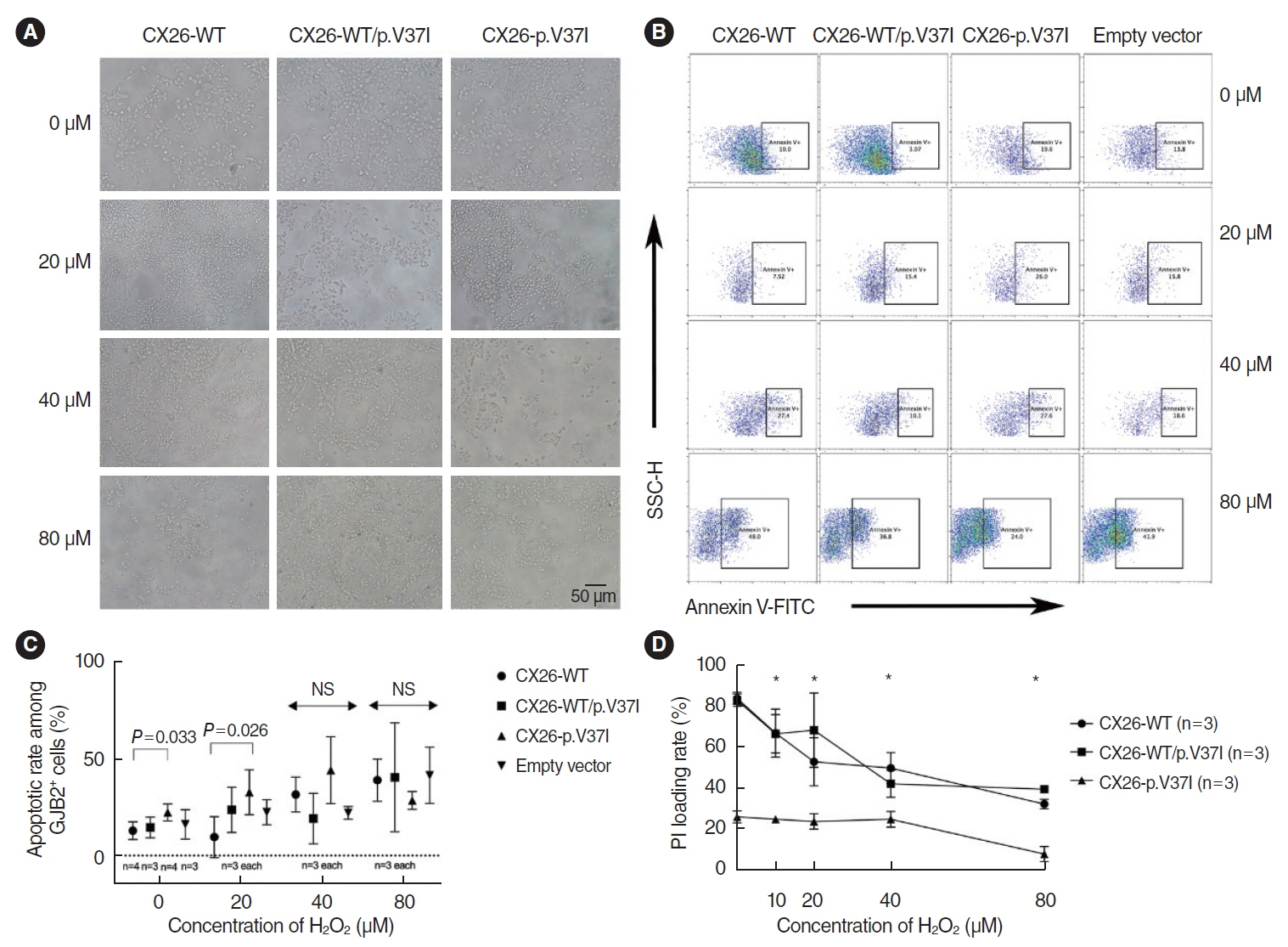
Fig. 5. (A) Representative results showing the morphological changes of HEK 293T cells with three haplotypes after H2O2 incubation. Scale bar=50 μm. (B) The tendency for apoptosis in HEK 293T cells expressing CX26-WT and/or CX26-p.V37I, treated with 20 μM to 80 μM H2O2. (C) Apoptosis rate. (D) Dynamic changes in biochemical hemichannel function in cells carrying three vectors. SSC-H, side scatter height; FITC, fluorescein isothiocyanate; NS, not significant. *P<0.05 when comparing the values in CX26-p.V37I with the values in CX26-WT or CX26-WT/p.V37I haplotypes.
Reference
-
1. Harris AL, Bevans CG. Exploring hemichannel permeability in vitro. Methods Mol Biol. 2001; 154:357–77.2. Dai P, Yu F, Han B, Liu X, Wang G, Li Q, et al. GJB2 mutation spectrum in 2,063 Chinese patients with nonsyndromic hearing impairment. J Transl Med. 2009; Apr. 7:26.3. Kenneson A, Van Naarden Braun K, Boyle C. GJB2 (connexin 26) variants and nonsyndromic sensorineural hearing loss: a HuGE review. Genet Med. 2002; Jul-Aug. 4(4):258–74.4. Chen K, Zong L, Liu M, Wang X, Zhou W, Zhan Y, et al. Developing regional genetic counseling for southern Chinese with nonsyndromic hearing impairment: a unique mutational spectrum. J Transl Med. 2014; Mar. 12:64.5. Tsukada K, Nishio S, Usami S, Deafness Gene Study Consortium. A large cohort study of GJB2 mutations in Japanese hearing loss patients. Clin Genet. 2010; Nov. 78(5):464–70.6. Oguchi T, Ohtsuka A, Hashimoto S, Oshima A, Abe S, Kobayashi Y, et al. Clinical features of patients with GJB2 (connexin 26) mutations: severity of hearing loss is correlated with genotypes and protein expression patterns. J Hum Genet. 2005; 50(2):76–83.7. Bruzzone R, Veronesi V, Gomes D, Bicego M, Duval N, Marlin S, et al. Loss-of-function and residual channel activity of connexin26 mutations associated with non-syndromic deafness. FEBS Lett. 2003; Jan. 533(1-3):79–88.8. Snoeckx RL, Huygen PL, Feldmann D, Marlin S, Denoyelle F, Waligora J, et al. GJB2 mutations and degree of hearing loss: a multicenter study. Am J Hum Genet. 2005; Dec. 77(6):945–57.9. Kelley PM, Harris DJ, Comer BC, Askew JW, Fowler T, Smith SD, et al. Novel mutations in the connexin 26 gene (GJB2) that cause autosomal recessive (DFNB1) hearing loss. Am J Hum Genet. 1998; Apr. 62(4):792–9.10. Gallant E, Francey L, Tsai EA, Berman M, Zhao Y, Fetting H, et al. Homozygosity for the V37I GJB2 mutation in fifteen probands with mild to moderate sensorineural hearing impairment: further confirmation of pathogenicity and haplotype analysis in Asian populations. Am J Med Genet A. 2013; Sep. 161A(9):2148–57.11. Jara O, Acuna R, Garcia IE, Maripillan J, Figueroa V, Saez JC, et al. Critical role of the first transmembrane domain of Cx26 in regulating oligomerization and function. Mol Biol Cell. 2012; Sep. 23(17):3299–311.12. Kim J, Jung J, Lee MG, Choi JY, Lee KA. Non-syndromic hearing loss caused by the dominant cis mutation R75Q with the recessive mutation V37I of the GJB2 (Connexin 26) gene. Exp Mol Med. 2015; Jun. 47(6):e169.13. Li L, Lu J, Tao Z, Huang Q, Chai Y, Li X, et al. The p.V37I exclusive genotype of GJB2: a genetic risk-indicator of postnatal permanent childhood hearing impairment. PLoS One. 2012; 7(5):e36621.14. Chen Y, Wang Z, Jiang Y, Lin Y, Wang X, Wang Z, et al. Biallelic p.V37I variant in GJB2 is associated with increasing incidence of hearing loss with age. Genet Med. 2022; Apr. 24(4):915–23.15. Choi SY, Lee KY, Kim HJ, Kim HK, Chang Q, Park HJ, et al. Functional evaluation of GJB2 variants in nonsyndromic hearing loss. Mol Med. 2011; May-Jun. 17(5-6):550–6.16. Choi SY, Park HJ, Lee KY, Dinh EH, Chang Q, Ahmad S, et al. Different functional consequences of two missense mutations in the GJB2 gene associated with non-syndromic hearing loss. Hum Mutat. 2009; Jul. 30(7):E716–27.17. Sun J, Ahmad S, Chen S, Tang W, Zhang Y, Chen P, et al. Cochlear gap junctions coassembled from Cx26 and 30 show faster intercellular Ca2+ signaling than homomeric counterparts. Am J Physiol Cell Physiol. 2005; Mar. 288(3):C613–23.18. Chen Y, Hu L, Wang X, Sun C, Lin X, Li L, et al. Characterization of a knock-in mouse model of the homozygous p.V37I variant in Gjb2. Sci Rep. 2016; Sep. 6:33279.19. Lin X, Li G, Zhang Y, Zhao J, Lu J, Gao Y, et al. Hearing consequences in Gjb2 knock-in mice: implications for human p.V37I mutation. Aging (Albany NY). 2019; Sep. 11(18):7416–41.20. Palmada M, Schmalisch K, Bohmer C, Schug N, Pfister M, Lang F, et al. Loss of function mutations of the GJB2 gene detected in patients with DFNB1-associated hearing impairment. Neurobiol Dis. 2006; Apr. 22(1):112–8.21. Hilgert N, Huentelman MJ, Thorburn AQ, Fransen E, Dieltjens N, Mueller-Malesinska M, et al. Phenotypic variability of patients homozygous for the GJB2 mutation 35delG cannot be explained by the influence of one major modifier gene. Eur J Hum Genet. 2009; Apr. 17(4):517–24.22. Hismi BO, Yilmaz ST, Incesulu A, Tekin M. Effects of GJB2 genotypes on the audiological phenotype: variability is present for all genotypes. Int J Pediatr Otorhinolaryngol. 2006; Oct. 70(10):1687–94.23. Liu LM, Liang C, Chen J, Fang S, Zhao HB. Cx26 heterozygous mutations cause hyperacusis-like hearing oversensitivity and increase susceptibility to noise. Sci Adv. 2023; Feb. 9(6):eadf4144.24. Fetoni AR, Zorzi V, Paciello F, Ziraldo G, Peres C, Raspa M, et al. Cx26 partial loss causes accelerated presbycusis by redox imbalance and dysregulation of Nfr2 pathway. Redox Biol. 2018; Oct. 19:301–17.25. Chiang YT, Lin PH, Lo MY, Chen HL, Lee CY, Tsai CY, et al. Genetic factors contribute to the phenotypic variability in GJB2-related hearing impairment. J Mol Diagn. 2023; Nov. 25(11):827–37.
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Genetics of Hearing Loss in North Iran Population: An Update of Spectrum and Frequency of GJB2 Mutations
- A Functional Study of Gap Junction in GJB2 Mutations Associated with Hereditary Hearing Loss
- Long Term Speech Perception Outcomes of Cochlear Implantation in Gap Junction Protein Beta 2 Related Hearing Loss
- A Case of Hystrix-Like Ichthyosis and Deafness Syndrome with a Rare Variant of Gap Junction Protein Beta 2 Gene Mutation
- Mutations in GJB2 as Major Causes of Autosomal Recessive Non-Syndromic Hearing Loss: First Report of c.299-300delAT Mutation in Kurdish Population of Iran
