Child Kidney Dis.
2024 Feb;28(1):8-15. 10.3339/ckd.24.005.
Navigating the landscape of clinical genetic testing: insights and challenges in rare disease diagnostics
- Affiliations
-
- 1Department of Genomic Medicine, Seoul National University Hospital, Seoul, Republic of Korea
- KMID: 2553175
- DOI: http://doi.org/10.3339/ckd.24.005
Abstract
- With the rapid evolution of diagnostic tools, particularly next-generation sequencing, the identification of genetic diseases, predominantly those with pediatric-onset, has significantly advanced. However, this progress presents challenges that span from selecting appropriate tests to the final interpretation of results. This review examines various genetic testing methodologies, each with specific indications and characteristics, emphasizing the importance of selecting the appropriate genetic test in clinical practice, taking into account factors like detection range, cost, turnaround time, and specificity of the clinical diagnosis. Interpretation of variants has become more challenging, often requiring further validation and significant resource allocation. Laboratories primarily classify variants based on the American College of Medical Genetics and Genomics and the Association for Clinical Genomic Science guidelines, however, this process has limitations. This review underscores the critical role of clinicians in matching patient phenotypes with reported genes/variants and considering additional factors such as variable expressivity, disease pleiotropy, and incomplete penetrance. These considerations should be aligned with specific gene-disease characteristics and segregation results based on an extended pedigree. In conclusion, this review aims to enhance understanding of the complexities of clinical genetic testing, advocating for a multidisciplinary approach to ensure accurate diagnosis and effective management of rare genetic diseases.
Keyword
Figure
-

Fig. 1. Conceptual representation of expressivity, pleiotropy, and penetrance in autosomal dominant genetic disorders. (A) A pedigree displaying an autosomal dominant genetic with varying levels of disease expressivity among family members. (B) A pedigree illustrating an autosomal dominant genetic disorder demonstrating disease pleiotropy within family members. (C) A pedigree of autosomal dominant genetic disorder exhibiting complete penetrance. (D) A pedigree of autosomal dominant genetic disorder exhibiting incomplete penetrance. Mut, mutation.
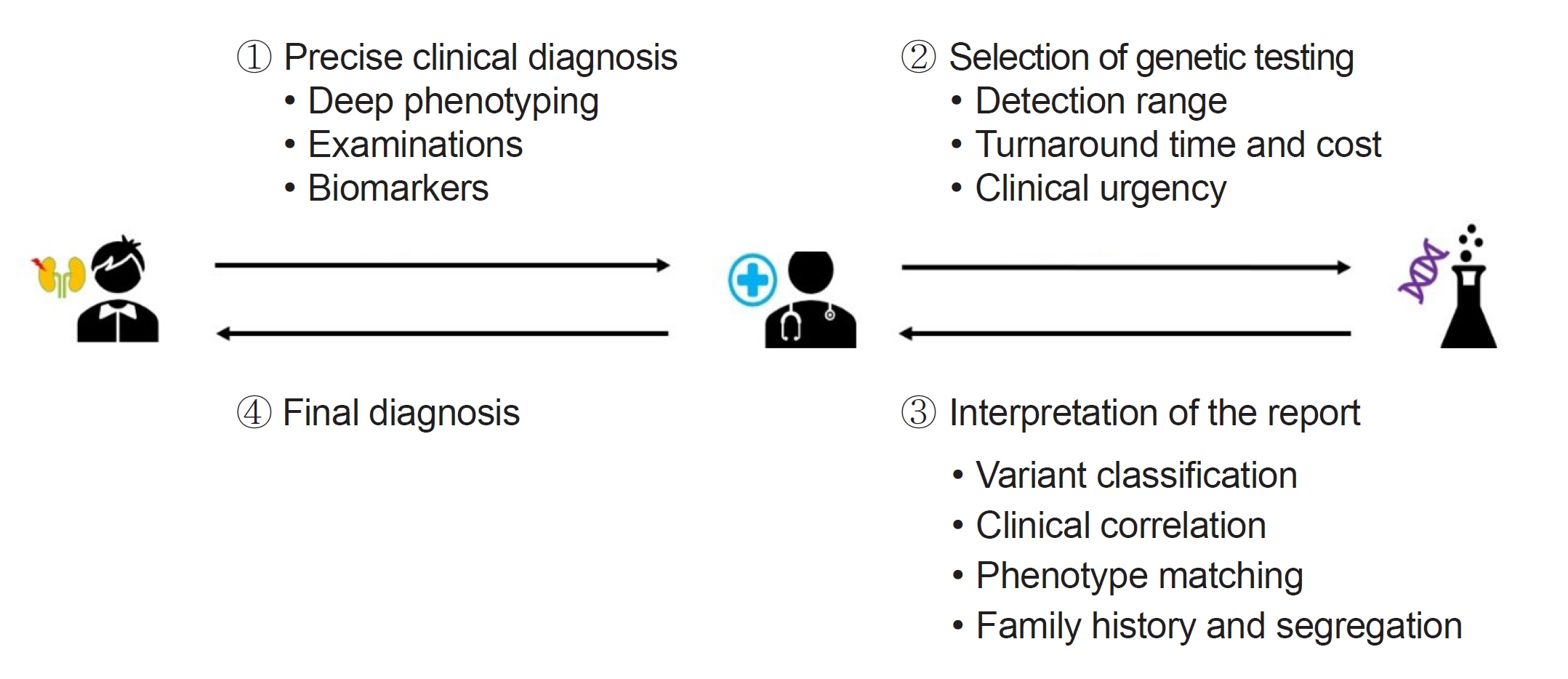
Fig. 2. Physician’s guide to genetic testing in rare diseases.
Reference
-
References
1. Boycott KM, Vanstone MR, Bulman DE, MacKenzie AE. Rare-disease genetics in the era of next-generation sequencing: discovery to translation. Nat Rev Genet. 2013; 14:681–91.
Article2. Orphanet activity report. Orphanet reports series/procedures [Internet]. Orphanet; 2022 [cited 2024 Jan 11]. Available from: https://www.orpha.net/consor/cgi-bin/Education_Home.php?lng=EN.3. Nguengang Wakap S, Lambert DM, Olry A, Rodwell C, Gueydan C, Lanneau V, et al. Estimating cumulative point prevalence of rare diseases: analysis of the Orphanet database. Eur J Hum Genet. 2020; 28:165–73.
Article4. Global Genes. RARE disease facts [Internet]. Global Genes; 2020 [cited 2024 Jan 11]. Available from: https://globalgenes.org/rare-facts/.5. Lalonde E, Rentas S, Lin F, Dulik MC, Skraban CM, Spinner NB. Genomic diagnosis for pediatric disorders: revolution and evolution. Front Pediatr. 2020; 8:373.
Article6. Yunis JJ. Mid-prophase human chromosomes: the attainment of 2000 bands. Hum Genet. 1981; 56:293–8.
Article7. Kallioniemi OP, Kallioniemi A, Sudar D, Rutovitz D, Gray JW, Waldman F, et al. Comparative genomic hybridization: a rapid new method for detecting and mapping DNA amplification in tumors. Semin Cancer Biol. 1993; 4:41–6.8. Solinas-Toldo S, Lampel S, Stilgenbauer S, Nickolenko J, Benner A, Dohner H, et al. Matrix-based comparative genomic hybridization: biochips to screen for genomic imbalances. Genes Chromosomes Cancer. 1997; 20:399–407.
Article9. Albertson DG, Pinkel D. Genomic microarrays in human genetic disease and cancer. Hum Mol Genet. 2003; 12 Spec No 2:R145–52.
Article10. Ahn JW, Bint S, Bergbaum A, Mann K, Hall RP, Ogilvie CM. Array CGH as a first line diagnostic test in place of karyotyping for postnatal referrals: results from four years’ clinical application for over 8,700 patients. Mol Cytogenet. 2013; 6:16.
Article11. Battaglia A, Doccini V, Bernardini L, Novelli A, Loddo S, Capalbo A, et al. Confirmation of chromosomal microarray as a first-tier clinical diagnostic test for individuals with developmental delay, intellectual disability, autism spectrum disorders and dysmorphic features. Eur J Paediatr Neurol. 2013; 17:589–99.
Article12. Stankiewicz P, Lupski JR. Structural variation in the human genome and its role in disease. Annu Rev Med. 2010; 61:437–55.
Article13. Vissers LE, de Vries BB, Veltman JA. Genomic microarrays in mental retardation: from copy number variation to gene, from research to diagnosis. J Med Genet. 2010; 47:289–97.
Article14. Rudkin GT, Stollar BD. High resolution detection of DNA-RNA hybrids in situ by indirect immunofluorescence. Nature. 1977; 265:472–3.
Article15. Iqbal MA, Ulmer C, Sakati N. Use of FISH technique in the diagnosis of chromosomal syndromes. East Mediterr Health J. 1999; 5:1218–24.
Article16. Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A. 1977; 74:5463–7.
Article17. Goossens M. The amplification of nucleotide sequences by PCR and the new technics for molecular diagnosis. Reprod Nutr Dev. 1990; Suppl 1:117s–124s.18. Ben-Ezra JM. Amplification methods in the molecular diagnosis of genetic diseases. Clin Lab Med. 1995; 15:795–815.
Article19. Erlich HA, Arnheim N. Genetic analysis using the polymerase chain reaction. Annu Rev Genet. 1992; 26:479–506.
Article20. Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002; 30:e57.
Article21. Stuppia L, Antonucci I, Palka G, Gatta V. Use of the MLPA assay in the molecular diagnosis of gene copy number alterations in human genetic diseases. Int J Mol Sci. 2012; 13:3245–76.
Article22. Borst M, Miller DM. DNA isolation and Southern analysis: a clinician's view. Am J Med Sci. 1990; 299:356–60.23. Southern EM. Detection of specific sequences among DNA fragments separated by gel electrophoresis. J Mol Biol. 1975; 98:503–17.
Article24. Reuter JA, Spacek DV, Snyder MP. High-throughput sequencing technologies. Mol Cell. 2015; 58:586–97.
Article25. Pettersson E, Lundeberg J, Ahmadian A. Generations of sequencing technologies. Genomics. 2009; 93:105–11.
Article26. Fernandez-Marmiesse A, Gouveia S, Couce ML. NGS technologies as a turning point in rare disease research, diagnosis and treatment. Curr Med Chem. 2018; 25:404–32.27. Chung CC, Hue SP, Ng NY, Doong PH; Hong Kong Genome Project, Chu AT, et al. Meta-analysis of the diagnostic and clinical utility of exome and genome sequencing in pediatric and adult patients with rare diseases across diverse populations. Genet Med. 2023; 25:100896.
Article28. Jais JP, Knebelmann B, Giatras I, De Marchi M, Rizzoni G, Renieri A, et al. X-linked Alport syndrome: natural history and genotype-phenotype correlations in girls and women belonging to 195 families: a “European Community Alport Syndrome Concerted Action” study. J Am Soc Nephrol. 2003; 14:2603–10.29. Bekheirnia MR, Reed B, Gregory MC, McFann K, Shamshirsaz AA, Masoumi A, et al. Genotype-phenotype correlation in X-linked Alport syndrome. J Am Soc Nephrol. 2010; 21:876–83.
Article30. Rao AN, Kavitha J, Koch M, Suresh Kumar V. Inborn errors of metabolism: review and data from a tertiary care center. Indian J Clin Biochem. 2009; 24:215–22.
Article31. Kim MJ, Kim SY, Lee JS, Kang S, Park LJ, Choi W, et al. Rapid targeted sequencing using dried blood spot samples for patients with suspected actionable genetic diseases. Ann Lab Med. 2023; 43:280–9.
Article32. Owen MJ, Niemi AK, Dimmock DP, Speziale M, Nespeca M, Chau KK, et al. Rapid sequencing-based diagnosis of thiamine metabolism dysfunction syndrome. N Engl J Med. 2021; 384:2159–61.
Article33. Wojcik MH, Callahan KP, Antoniou A, Del Rosario MC, Brunelli L, ElHassan NO, et al. Provision and availability of genomic medicine services in level IV neonatal intensive care units. Genet Med. 2023; 25:100926.
Article34. Incerti D, Xu XM, Chou JW, Gonzaludo N, Belmont JW, Schroeder BE. Cost-effectiveness of genome sequencing for diagnosing patients with undiagnosed rare genetic diseases. Genet Med. 2022; 24:109–18.
Article35. Runheim H, Pettersson M, Hammarsjo A, Nordgren A, Henriksson M, Lindstrand A, et al. The cost-effectiveness of whole genome sequencing in neurodevelopmental disorders. Sci Rep. 2023; 13:6904.
Article36. Yeung A, Tan NB, Tan TY, Stark Z, Brown N, Hunter MF, et al. A cost-effectiveness analysis of genomic sequencing in a prospective versus historical cohort of complex pediatric patients. Genet Med. 2020; 22:1986–93.
Article37. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015; 17:405–24.
Article38. Ellard S, Baple EL, Callaway A, Berry I, Forrester N, Turnbull C, et al. ACGS best practice guidelines for variant classification in Rare Disease 2020 [Internet]. Association for Clinical Genomic Science; 2020 [cited 2024 Jan 11]. Available from: https://www.acgs.uk.com/media/11631/uk-practice-guidelines-for-variant-classification-v4-01-2020.pdf.39. Amendola LM, Jarvik GP, Leo MC, McLaughlin HM, Akkari Y, Amaral MD, et al. Performance of ACMG-AMP variant-interpretation guidelines among nine laboratories in the clinical sequencing exploratory research consortium. Am J Hum Genet. 2016; 98:1067–76.
Article40. Inoue Y, Machida O, Kita Y, Yamamoto T. Need for revision of the ACMG/AMP guidelines for interpretation of X-linked variants. Intractable Rare Dis Res. 2022; 11:120–4.
Article41. Patel MJ, DiStefano MT, Oza AM, Hughes MY, Wilcox EH, Hemphill SE, et al. Disease-specific ACMG/AMP guidelines improve sequence variant interpretation for hearing loss. Genet Med. 2021; 23:2208–12.
Article42. Strande NT, Brnich SE, Roman TS, Berg JS. Navigating the nuances of clinical sequence variant interpretation in Mendelian disease. Genet Med. 2018; 20:918–26.
Article43. Gelb BD, Cave H, Dillon MW, Gripp KW, Lee JA, Mason-Suares H, et al. ClinGen’s RASopathy Expert Panel consensus methods for variant interpretation. Genet Med. 2018; 20:1334–45.
Article44. Savige J, Storey H, Watson E, Hertz JM, Deltas C, Renieri A, et al. Consensus statement on standards and guidelines for the molecular diagnostics of Alport syndrome: refining the ACMG criteria. Eur J Hum Genet. 2021; 29:1186–97.
Article45. Chen E, Facio FM, Aradhya KW, Rojahn S, Hatchell KE, Aguilar S, et al. Rates and classification of variants of uncertain significance in hereditary disease genetic testing. JAMA Netw Open. 2023; 6:e2339571.
Article46. Burke W, Parens E, Chung WK, Berger SM, Appelbaum PS. The challenge of genetic variants of uncertain clinical significance : a narrative review. Ann Intern Med. 2022; 175:994–1000.
Article47. Johnson B, Ouyang K, Frank L, Truty R, Rojahn S, Morales A, et al. Systematic use of phenotype evidence in clinical genetic testing reduces the frequency of variants of uncertain significance. Am J Med Genet A. 2022; 188:2642–51.
Article48. Nykamp K, Anderson M, Powers M, Garcia J, Herrera B, Ho YY, et al. Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria. Genet Med. 2017; 19:1105–17.
Article49. Kingdom R, Wright CF. Incomplete penetrance and variable expressivity: from clinical studies to population cohorts. Front Genet. 2022; 13:920390.
Article50. Rossetti S, Kubly VJ, Consugar MB, Hopp K, Roy S, Horsley SW, et al. Incompletely penetrant PKD1 alleles suggest a role for gene dosage in cyst initiation in polycystic kidney disease. Kidney Int. 2009; 75:848–55.
Article51. Benson PF, Barbarik A, Brown SP, Mann TP. GM1-generalized gangliosidosis variant with cardiomegaly. Postgrad Med J. 1976; 52:159–65.
Article52. Arbisser AI, Donnelly KA, Scott CI Jr, DiFerrante N, Singh J, Stevenson RE, et al. Morquio-like syndrome with beta galactosidase deficiency and normal hexosamine sulfatase activity: mucopolysacchariodosis IVB. Am J Med Genet. 1977; 1:195–205.
Article53. Vytopil M, Ricci E, Dello Russo A, Hanisch F, Neudecker S, Zierz S, et al. Frequent low penetrance mutations in the Lamin A/C gene, causing Emery Dreifuss muscular dystrophy. Neuromuscul Disord. 2002; 12:958–63.
Article54. Burdon KP, Wirth MG, Mackey DA, Russell-Eggitt IM, Craig JE, Elder JE, et al. A novel mutation in the Connexin 46 gene causes autosomal dominant congenital cataract with incomplete penetrance. J Med Genet. 2004; 41:e106.
Article55. Shawky RM. Reduced penetrance in human inherited disease. Egypt J Medl Hum Genet. 2014; 15:103–11.
Article56. Pereira R, Halford K, Sokolov BP, Khillan JS, Prockop DJ. Phenotypic variability and incomplete penetrance of spontaneous fractures in an inbred strain of transgenic mice expressing a mutated collagen gene (COL1A1). J Clin Invest. 1994; 93:1765–9.
Article57. Van Dijk FS, Sillence DO. Osteogenesis imperfecta: clinical diagnosis, nomenclature and severity assessment. Am J Med Genet A. 2014; 164A:1470–81.
Article58. Ellingford JM, Hufnagel RB, Arno G. Phenotype and genotype correlations in inherited retinal diseases: population-guided variant interpretation, variable expressivity and incomplete penetrance. Genes (Basel). 2020; 11:1274.
Article59. Vujic M, Heyer CM, Ars E, Hopp K, Markoff A, Orndal C, et al. Incompletely penetrant PKD1 alleles mimic the renal manifestations of ARPKD. J Am Soc Nephrol. 2010; 21:1097–102.
Article60. Stein Q, Westemeyer M, Darwish T, Pitman T, Hager M, Tabriziani H, et al. Genetic counseling in kidney disease: a perspective. Kidney Med. 2023; 5:100668.
Article61. Burger J, Fonknechten N, Hoeltzenbein M, Neumann L, Bratanoff E, Hazan J, et al. Hereditary spastic paraplegia caused by mutations in the SPG4 gene. Eur J Hum Genet. 2000; 8:771–6.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Navigating the Treatment Landscape for Widespread Superficial Esophageal Squamous Cell Neoplasia
- Clinical Genetic Testing in Children with Kidney Disease
- Genetics of Cerebral Palsy: Diagnosis, Differential Diagnosis, and Beyond
- Genomic testing for germline predisposition to hematologic malignancies
- Direct-to-consumer genetic testing
