Genetic Analysis and Clinical Characteristics of Hereditary Pheochromocytoma and Paraganglioma Syndrome in Korean Population
- Affiliations
-
- 1Yonsei University College of Medicine, Seoul, Korea
- 2Department of Internal Medicine, Severance Hospital, Endocrine Research Institute, Yonsei University College of Medicine, Seoul, Korea
- 3Department of Internal Medicine, Korea University College of Medicine, Seoul, Korea
- 4Department of Laboratory Medicine, Yonsei University College of Medicine, Seoul, Korea
- 5Thyroid-Endocrine Surgery Division, Department of Surgery, Yonsei University College of Medicine, Seoul, Korea
- KMID: 2511013
- DOI: http://doi.org/10.3803/EnM.2020.683
Abstract
- Background
Pheochromocytoma and paragangliomas (PPGL) are hereditary in approximately 30% to 40% cases. With the advancement of genetic analysis techniques, including next-generation sequencing (NGS), there were attempts to classify PPGL into molecular clusters. With NGS being applied to clinical settings recently, we aimed to review the results of genetic analysis, including NGS, and investigate the association with clinical characteristics in Korean PPGL patients.
Methods
We reviewed the medical records of PPGL patients who visited Severance hospital from 2006 to 2019. We documented the clinical phenotype of those who underwent targeted NGS or had known germline mutations of related genes.
Results
Among 57 PPGL patients, we found 28 pathogenic germline mutations of susceptibility genes. Before the targeted NGS was implemented, only obvious syndromic feature lead to the Sanger sequencing for the specific genes. Therefore, for the exact prevalence, only patients after the year 2017, when targeted NGS was added, were included (n=43). The positive germline mutations were found in 14 patients; thus, the incidence rate is 32.6%. Patients with germline mutations had a higher likelihood of family history. There were significant differences in the type of PPGLs, percentage of family history, metastasis rate, presence of other tumors, and biochemical profile among three molecular clusters: pseudohypoxic tricarboxylic acid cycle-related, pseudohypoxic von Hippel-Lindau (VHL)/endothelial PAS domain-containing protein 1-related, and kinase-signaling group. Germline mutations were identified in seven PPGL-related genes (SDHB, RET, VHL, NF1, MAX, SDHA, and SDHD).
Conclusion
We report the expected prevalence of germline mutations in Korean PPGL patients. NGS is a useful and accessible tool for genetic analysis in patients with PPGLs, and further research on molecular classification is needed for precise management.
Keyword
Figure
-
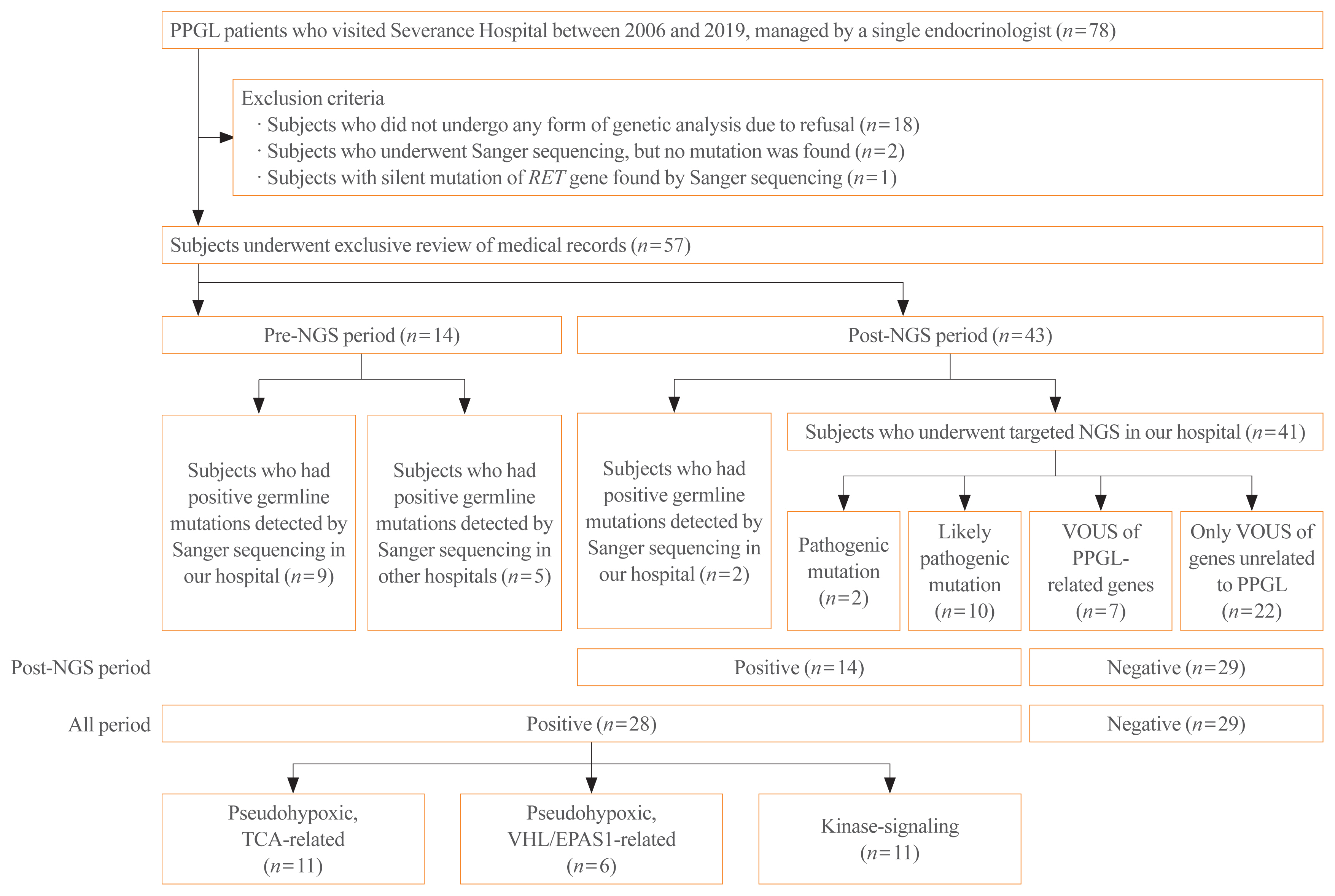
Fig. 1 Study flow chart. PPGL, pheochromocytoma and paraganglioma; RET, rearranged during transformation; NGS, next-generation sequencing; VOUS, variant of unknown significance; TCA, tricarboxylic acid; VHL, von Hippel-Lindau; EPAS1, endothelial PAS domain-containing protein 1.

Fig. 2 The frequency of germline mutations in susceptibility genes related to pheochromocytomas and paragangliomas detected in our study. SDH, succinate dehydrogenase; RET, rearranged during transformation; VHL, von Hippel-Lindau; NF1, neurofibromatosis1; MAX, Myc-associated protein X.

Fig. 3 Overview of mutation distribution in pheochromocytoma and paraganglioma patients of this study.
Cited by 1 articles
-
Diagnosis for Pheochromocytoma and Paraganglioma: A Joint Position Statement of the Korean Pheochromocytoma and Paraganglioma Task Force
Eu Jeong Ku, Kyoung Jin Kim, Jung Hee Kim, Mi Kyung Kim, Chang Ho Ahn, Kyung Ae Lee, Seung Hun Lee, You-Bin Lee, Kyeong Hye Park, Yun Mi Choi, Namki Hong, A Ram Hong, Sang-Wook Kang, Byung Kwan Park, Moon-Woo Seong, Myungshin Kim, Kyeong Cheon Jung, Chan Kwon Jung, Young Seok Cho, Jin Chul Paeng, Jae Hyeon Kim, Ohk-Hyun Ryu, Yumie Rhee, Chong Hwa Kim, Eun Jig Lee
Endocrinol Metab. 2021;36(2):322-338. doi: 10.3803/EnM.2020.908.
Reference
-
1. Lam AK. Update on adrenal tumours in 2017 World Health Organization (WHO) of endocrine tumours. Endocr Pathol. 2017; 28:213–27.
Article2. McNichol AM. Differential diagnosis of pheochromocytomas and paragangliomas. Endocr Pathol. 2001; 12:407–15.
Article3. Karagiannis A, Mikhailidis DP, Athyros VG, Harsoulis F. Pheochromocytoma: an update on genetics and management. Endocr Relat Cancer. 2007; 14:935–56.
Article4. Beard CM, Sheps SG, Kurland LT, Carney JA, Lie JT. Occurrence of pheochromocytoma in Rochester, Minnesota, 1950 through 1979. Mayo Clin Proc. 1983; 58:802–4.5. Johnson MH, Cavallo JA, Figenshau RS. Malignant and metastatic pheochromocytoma: case report and review of the literature. Urol Case Rep. 2014; 2:139–41.
Article6. Bravo EL, Gifford RW Jr. Current concepts: pheochromocytoma: diagnosis, localization and management. N Engl J Med. 1984; 311:1298–303.
Article7. Manger WM, Gifford RW Jr, Hoffman BB. Pheochromocytoma: a clinical and experimental overview. Curr Probl Cancer. 1985; 9:1–89.8. Favier J, Amar L, Gimenez-Roqueplo AP. Paraganglioma and phaeochromocytoma: from genetics to personalized medicine. Nat Rev Endocrinol. 2015; 11:101–11.
Article9. Dahia PL. Pheochromocytoma and paraganglioma pathogenesis: learning from genetic heterogeneity. Nat Rev Cancer. 2014; 14:108–19.
Article10. Mannelli M, Castellano M, Schiavi F, Filetti S, Giacche M, Mori L, et al. Clinically guided genetic screening in a large cohort of Italian patients with pheochromocytomas and/or functional or nonfunctional paragangliomas. J Clin Endocrinol Metab. 2009; 94:1541–7.
Article11. Metzker ML. Sequencing technologies: the next generation. Nat Rev Genet. 2010; 11:31–46.12. Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 2000; 287:848–51.
Article13. Nakamura K, Yamaki M, Sarada M, Nakayama S, Vibat CR, Gennis RB, et al. Two hydrophobic subunits are essential for the heme b ligation and functional assembly of complex II (succinate-ubiquinone oxidoreductase) from Escherichia coli. J Biol Chem. 1996; 271:521–7.
Article14. Luchetti A, Walsh D, Rodger F, Clark G, Martin T, Irving R, et al. Profiling of somatic mutations in phaeochromocytoma and paraganglioma by targeted next generation sequencing analysis. Int J Endocrinol. 2015; 2015:138573.
Article15. Crona J, Taieb D, Pacak K. New perspectives on pheochromocytoma and paraganglioma: toward a molecular classification. Endocr Rev. 2017; 38:489–515.
Article16. Lenders JW, Duh QY, Eisenhofer G, Gimenez-Roqueplo AP, Grebe SK, Murad MH, et al. Pheochromocytoma and paraganglioma: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2014; 99:1915–42.
Article17. NGS in PPGL (NGSnPPGL) Study Group. Toledo RA, Burnichon N, Cascon A, Benn DE, Bayley JP, et al. Consensus statement on next-generation-sequencing-based diagnostic testing of hereditary phaeochromocytomas and paragangliomas. Nat Rev Endocrinol. 2017; 13:233–47.
Article18. Cascon A, Pita G, Burnichon N, Landa I, Lopez-Jimenez E, Montero-Conde C, et al. Genetics of pheochromocytoma and paraganglioma in Spanish patients. J Clin Endocrinol Metab. 2009; 94:1701–5.
Article19. Taschner PE, Jansen JC, Baysal BE, Bosch A, Rosenberg EH, Brocker-Vriends AH, et al. Nearly all hereditary paragangliomas in the Netherlands are caused by two founder mutations in the SDHD gene. Genes Chromosomes Cancer. 2001; 31:274–81.20. Kim KH, Chung JS, Kim WT, Oh CK, Chae YB, Yu HS, et al. Clinical experiences of pheochromocytoma in Korea. Yonsei Med J. 2011; 52:45–50.
Article21. Kim JH, Seong MW, Lee KE, Choi HJ, Ku EJ, Bae JH, et al. Germline mutations and genotype-phenotype correlations in patients with apparently sporadic pheochromocytoma/paraganglioma in Korea. Clin Genet. 2014; 86:482–6.
Article22. Plouin PF, Amar L, Dekkers OM, Fassnacht M, Gimenez-Roqueplo AP, Lenders JW, et al. European Society of Endocrinology Clinical Practice Guideline for long-term follow-up of patients operated on for a phaeochromocytoma or a paraganglioma. Eur J Endocrinol. 2016; 174:G1–10.
Article23. Jochmanova I, Lazurova I. Diagnostika a manazment metastatickeho feochromocytomu a paragangliomu. Diagnosis and management of metastatic pheochromocytoma and paraganglioma. Vnitr Lek. 2017; 63:580–8.24. Chrisoulidou A, Kaltsas G, Ilias I, Grossman AB. The diagnosis and management of malignant phaeochromocytoma and paraganglioma. Endocr Relat Cancer. 2007; 14:569–85.
Article25. Amar L, Fassnacht M, Gimenez-Roqueplo AP, Januszewicz A, Prejbisz A, Timmers H, et al. Long-term postoperative follow-up in patients with apparently benign pheochromocytoma and paraganglioma. Horm Metab Res. 2012; 44:385–9.
Article26. Asari R, Scheuba C, Kaczirek K, Niederle B. Estimated risk of pheochromocytoma recurrence after adrenal-sparing surgery in patients with multiple endocrine neoplasia type 2A. Arch Surg. 2006; 141:1199–205.
Article27. Pacak K, Wimalawansa SJ. Pheochromocytoma and paraganglioma. Endocr Pract. 2015; 21:406–12.
Article28. Li MM, Datto M, Duncavage EJ, Kulkarni S, Lindeman NI, Roy S, et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: a joint consensus recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn. 2017; 19:4–23.
Article29. DeLelis RL, Lloyd RV, Heitz PU, Eng C. Pathology and genetics of tumours of endocrine organs (IARC/World Health Organization Classification of Tumours). Lyon: IARC Press;2004.30. Rijken JA, Niemeijer ND, Jonker MA, Eijkelenkamp K, Jansen JC, van Berkel A, et al. The penetrance of paraganglioma and pheochromocytoma in SDHB germline mutation carriers. Clin Genet. 2018; 93:60–6.
Article31. Welander J, Soderkvist P, Gimm O. Genetics and clinical characteristics of hereditary pheochromocytomas and paragangliomas. Endocr Relat Cancer. 2011; 18:R253–76.
Article32. Neumann HP, Bausch B, McWhinney SR, Bender BU, Gimm O, Franke G, et al. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med. 2002; 346:1459–66.
Article33. Adler JT, Meyer-Rochow GY, Chen H, Benn DE, Robinson BG, Sippel RS, et al. Pheochromocytoma: current approaches and future directions. Oncologist. 2008; 13:779–93.
Article34. Amar L, Baudin E, Burnichon N, Peyrard S, Silvera S, Bertherat J, et al. Succinate dehydrogenase B gene mutations predict survival in patients with malignant pheochromocytomas or paragangliomas. J Clin Endocrinol Metab. 2007; 92:3822–8.
Article35. Eisenhofer G, Lenders JW, Siegert G, Bornstein SR, Friberg P, Milosevic D, et al. Plasma methoxytyramine: a novel biomarker of metastatic pheochromocytoma and paraganglioma in relation to established risk factors of tumour size, location and SDHB mutation status. Eur J Cancer. 2012; 48:1739–49.
Article36. Gimenez-Roqueplo AP, Favier J, Rustin P, Rieubland C, Crespin M, Nau V, et al. Mutations in the SDHB gene are associated with extra-adrenal and/or malignant phaeochromocytomas. Cancer Res. 2003; 63:5615–21.37. Chen X, Hu L, Liu C, Ni G, Zhang Y. Tumor characteristics and surgical outcome in incidentally discovered pheochromocytomas and paragangliomas. Endocr Connect. 2018; 7:1142–9.
Article38. Mariani-Costantini R. Paraganglioma: a multidisciplinary approach. Brisbane: Codon Publications;2019.39. Hoffman-Andrews L. The known unknown: the challenges of genetic variants of uncertain significance in clinical practice. J Law Biosci. 2018; 4:648–57.
Article40. Geli J, Kiss N, Karimi M, Lee JJ, Backdahl M, Ekstrom TJ, et al. Global and regional CpG methylation in pheochromocytomas and abdominal paragangliomas: association to malignant behavior. Clin Cancer Res. 2008; 14:2551–9.
Article41. Margetts CD, Astuti D, Gentle DC, Cooper WN, Cascon A, Catchpoole D, et al. Epigenetic analysis of HIC1, CASP8, FLIP, TSP1, DCR1, DCR2, DR4, DR5, KvDMR1, H19 and preferential 11p15.5 maternal-allele loss in von Hippel-Lindau and sporadic phaeochromocytomas. Endocr Relat Cancer. 2005; 12:161–72.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Clinical Analysis of Pheochromocytoma and Abdominal Paragangliomas
- Bilateral adrenal pheochromocytoma with a germline L790F mutation in the RET oncogene
- A Brief Overview of the Epidemiology of Pheochromocytoma and Paraganglioma in Korea
- Non-Functional Retroperitoneal Paraganglioma Mimicking an Ovary Mass: A Case Report
- A Case of Paraganglioma Presenting as Stroke
