Neonatal Med.
2020 Aug;27(3):141-146. 10.5385/nm.2020.27.3.141.
Case of a Male Newborn with Incontinentia Pigmenti Initially Misdiagnosed as a Recurrent Skin Infection
- Affiliations
-
- 1Departments of Pediatrics , Presbyterian Medical Center, Jeonju, Korea
- 2Departments of Dermatology, Presbyterian Medical Center, Jeonju, Korea
- KMID: 2505665
- DOI: http://doi.org/10.5385/nm.2020.27.3.141
Abstract
- Incontinentia pigmenti (IP), also known as Bloch-Sulzberger syndrome, is a rare X-linked dominant disorder that is generally lethal to males and almost always leads to death in utero. This disorder is a genodermatosis with characteristic cutaneous lesions and manifestations affecting the eyes, teeth, hair, and central nervous system. Genodermatosis is a hereditary disease caused by mutations in the nuclear factor-kappa B essential modulator gene mapped to chromosome Xq28. This gene encodes a variety of cytokines and chemokine regulators and is indispensable for protecting cells from tumor necrosis factor-induced apoptosis. Here we describe a case of male newborn with vesiculobullous cutaneous lesions over the left thigh and leg. We first considered the cutaneous lesions a skin infection, as they improved with intravenous antibiotics. However, recurrence and the need for repeated hospitali zations made us consider the differential diagnosis of IP, for which we performed a skin biopsy and chromosome analysis. The histology results were compatible with IP, that is, eosinophilic infiltration in the dermis and epidermis, and individual cell dyskeratinization. The chromosome analysis result was a normal 46, XY karyotype. Here we report the case of a male newborn with IP that manifested as multiple vesiculobullous skin lesions and was initially misdiagnosed as a recurrent skin infection.
Figure
-
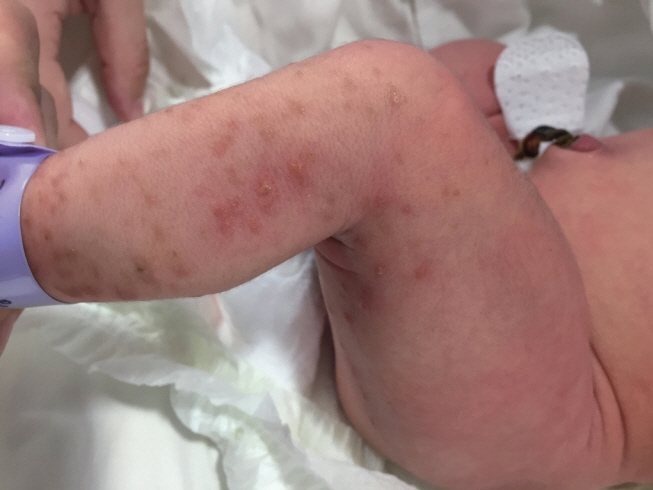
Figure 1. On the 4th day, clustered erythematous vesicles and light brownish macules are visible along the Blaschko line on the left extremities.n
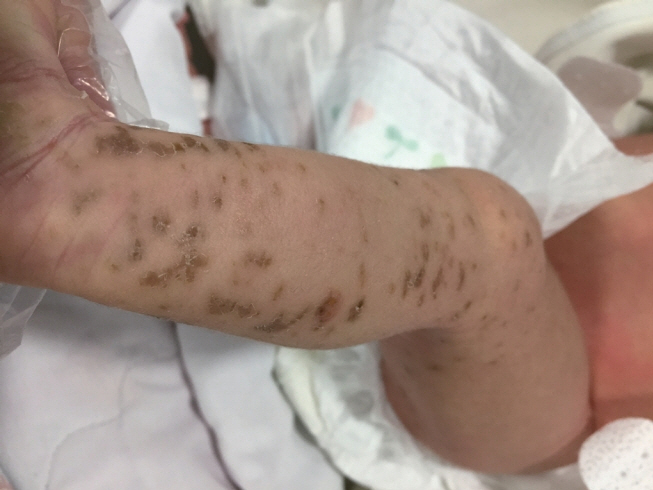
Figure 2. On the 11th day, blistered skin is replaced by crusts and dark brown colored pigmented macules on the left extremities.
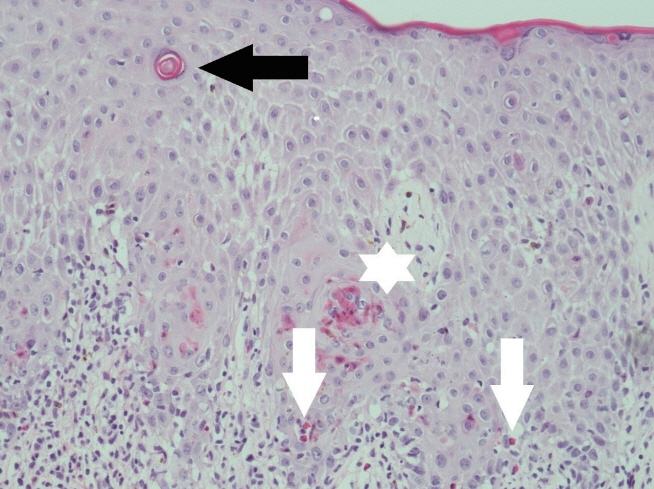
Figure 3. Histologic features of eosinophilic infiltration (white arrows) in the dermis and epidermis accompanying the vesicles and spongiosis, and individual cell dyskeratinization (asterisk) with keratin pearl (black arrow) formation (H&E stain, ×200).
Reference
-
1. Orphanet Report Series: rare diseases collection. Prevalence and incidence of rare diseases: bibliographic data [Internet]. Paris: Orphanet;2020. [cited 2020 Aug 12]. Available from: https://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_alphabetical_list.pdf.2. Fusco F, Paciolla M, Napolitano F, Pescatore A, D'Addario I, Bal E, et al. Genomic architecture at the Incontinentia Pigmenti locus favours de novo pathological alleles through different mechanisms. Hum Mol Genet. 2012; 21:1260–71.3. Berlin AL, Paller AS, Chan LS. Incontinentia pigmenti: a review and update on the molecular basis of pathophysiology. J Am Acad Dermatol. 2002; 47:169–87.4. Cammarata-Scalisi F, Fusco F, Ursini MV. Incontinentia pigmenti. Actas Dermosifiliogr. 2019; 110:273–8.5. Sulzberger MB. Uber eine bisher nicht beschriebene congenitale Pigmentanomalie [About a previously udescribed congenital pigment anomaly]. Arch Dermatol Syph (Berl). 1928; 154:19–32.6. Greene-Roethke C. Incontinentia pigmenti: a summary review of this rare ectodermal dysplasia with neurologic manifestations, including treatment protocols. J Pediatr Health Care. 2017; 31:e45–52.7. Fusco F, Valente V, Fergola D, Pescatore A, Lioi MB, Ursini MV. The incontinentia pigmenti genetic biobank: study design and cohort profile to facilitate research into a rare disease worldwide. Eur J Hum Genet. 2019; 27:1509–18.8. Sebban H, Courtois G. NF-kappaB and inflammation in genetic disease. Biochem Pharmacol. 2006; 72:1153–60.9. Carney RG. Incontinentia pigmenti: a world statistical analysis. Arch Dermatol. 1976; 112:535–42.10. Kenwrick S, Woffendin H, Jakins T, Shuttleworth SG, Mayer E, Greenhalgh L, et al. Survival of male patients with incontinentia pigmenti carrying a lethal mutation can be explained by somatic mosaicism or Klinefelter syndrome. Am J Hum Genet. 2001; 69:1210–7.11. Landy SJ, Donnai D. Incontinentia pigmenti (Bloch-Sulzberger syndrome). J Med Genet. 1993; 30:53–9.12. Minic S, Trpinac D, Obradovic M. Incontinentia pigmenti diagnostic criteria update. Clin Genet. 2014; 85:536–42.13. Rosser T. Neurocutaneous disorders. Continuum (Minneap Minn). 2018; 24:96–129.14. Park SS, Kook HI. A case of incontinentia pigmenti (Bloch-Sulzberger type) in male. Korean J Dermatol. 1982; 20:487–491.15. Kim BJ, Shin HS, Won CH, Lee JH, Kim KH, Kim MN, et al. Incontinentia pigmenti: clinical observation of 40 Korean cases. J Korean Med Sci. 2006; 21:474–7.16. Ocana Jaramillo S, Del Boz J, Vera Casano A. Incontinentia pigmenti: a descriptive study of experience in two different hospitals. An Pediatr (Barc). 2020; 92:3–12.17. Kim TH, Choi YJ, Park HK, Kim CR, Lee HJ. A case of incontinentia pigmenti with multiple brain infarction. Neonatal Med. 2013; 20:228–32.18. Cho MJ, Shin SM, Moon HK. A case of incontinentia pigmenti developed in a male newborn infant. Yeungnam Univ J Med. 1998; 15:381–90.19. Kim BJ, Lee DH, Shin HS, Won CH, Lee JH, Kwon OS. Incontinentia pigmenti in a male infant. Korean J Dermatol. 2006; 44:624–6.20. Song JY, Na CH, Chung BS, Choi KC, Shin BS. A case of a surviving male infant with incontinentia pigmenti. Ann Dermatol. 2008; 20:134–7.21. Hadj-Rabia S, Froidevaux D, Bodak N, Hamel-Teillac D, Smahi A, Touil Y, et al. Clinical study of 40 cases of incontinentia pigmenti. Arch Dermatol. 2003; 139:1163–70.22. Pacheco TR, Levy M, Collyer JC, de Parra NP, Parra CA, Garay M, et al. Incontinentia pigmenti in male patients. J Am Acad Dermatol. 2006; 55:251–5.23. Fusco F, Paciolla M, Conte MI, Pescatore A, Esposito E, Mirabelli P, et al. Incontinentia pigmenti: report on data from 2000 to 2013. Orphanet J Rare Dis. 2014; 9:93.
