Yonsei Med J.
2019 Dec;60(12):1209-1215. 10.3349/ymj.2019.60.12.1209.
Diagnostic Challenges Associated with GLUT1 Deficiency: Phenotypic Variabilities and Evolving Clinical Features
- Affiliations
-
- 1Department of Pediatrics, Seoul National University Children's Hospital, Seoul National University College of Medicine, Seoul, Korea. murimchoi@snu.ac.kr, chaeped1@snu.ac.kr
- 2Pediatric Clinical Neuroscience Center, Seoul National University Children's Hospital, Seoul National University College of Medicine, Seoul, Korea.
- 3Department of Pediatrics, Department of Genome Medicine and Science, Gil Medical Center, Gachon University College of Medicine, Incheon, Korea.
- 4Department of Biomedical Sciences, Seoul National University College of Medicine, Seoul, Korea.
- KMID: 2463801
- DOI: http://doi.org/10.3349/ymj.2019.60.12.1209
Abstract
- GLUT1 deficiency is a rare neurometabolic disorder that can be effectively treated with ketogenic diet. However, this condition is underdiagnosed due to its nonspecific, overlapping, and evolving symptoms with age. We retrospectively reviewed the clinical course of nine patients diagnosed with GLUT1 deficiency, based on SLC2A1 mutations and/or glucose concentration in cerebrospinal fluid. The patients included eight boys and one girl who initially presented with seizures (44%, 4/9) or delayed development (44%, 4/9) before 2 years of age, except for one patient who presented with apnea as a neonate. Over the clinical course, all of the children developed seizures of the mixed type, including absence seizures and generalized tonic-clonic seizures. About half (56%, 5/9) showed movement disorders such as ataxia, dystonia, or dyskinesia. We observed an evolution of phenotype over time, although this was not uniform across all patients. Only one child had microcephaly. In five patients, ketogenic diet was effective in reducing seizures and movement symptoms, and the patients exhibited subjective improvement in cognitive function. Diagnosing GLUT1 deficiency can be challenging due to the phenotypic variability and evolution. A high index of clinical suspicion in pediatric and even older patients with epilepsy or movement disorders is key to the early diagnosis and treatment, which can improve the patient's quality of life.
MeSH Terms
Figure
-
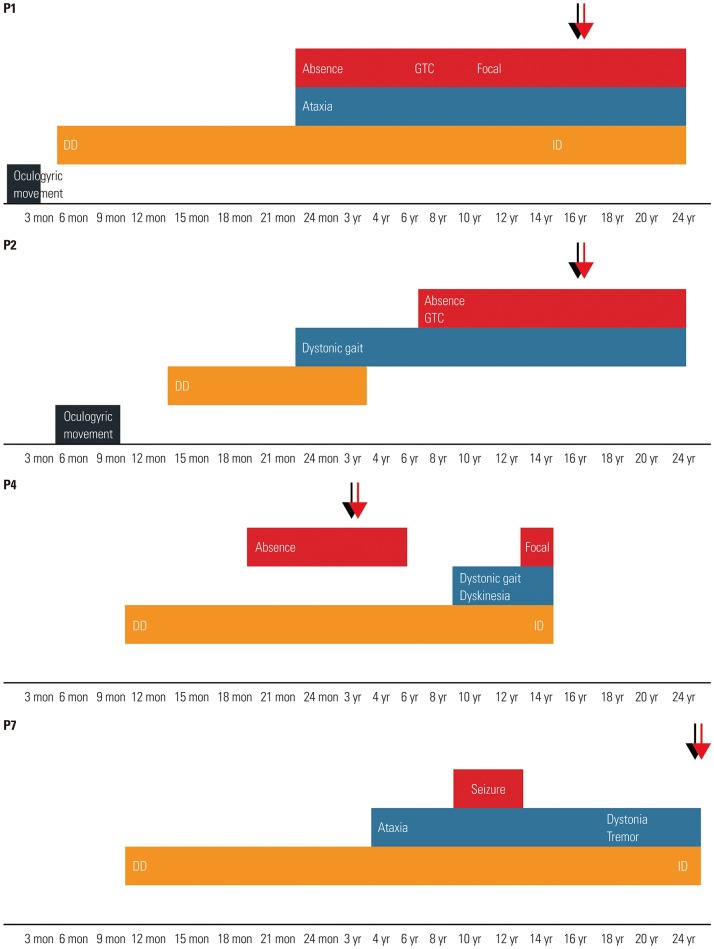
Fig. 1 The graph represents evolving phenotypes of four patients (Patients 1, 2, 4, and 7). Lower axis of the graph represents patient's age. Red and light blue represent epilepsy and movement disorders, respectively. Orange represents developmental delay (DD) and intellectual disability (ID). Black represents other atypical symptoms. Black arrows represent age at diagnosis, and red arrows denote age during treatment (ketogenic diet). GTC, generalized tonic–clonic.
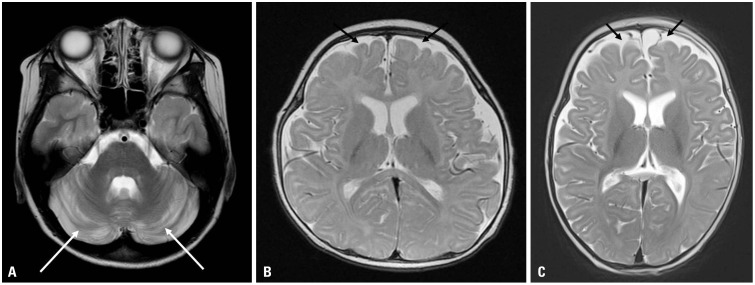
Fig. 2 Brain MRI axial T2-weighted images of three patients show cerebellar atrophy (white arrows) at 12 years of age in Patient 3 (A), and slightly delayed myelination with prominent extra-axial space widening (black arrows) in Patients 5 and 6 at 5 months of age (B and C).
Reference
-
1. Gras D, Roze E, Caillet S, Méneret A, Doummar D, Billette de Villemeur T, et al. GLUT1 deficiency syndrome: an update. Rev Neurol (Paris). 2014; 170:91–99. PMID: 24269118.
Article2. De Vivo DC, Trifiletti RR, Jacobson RI, Ronen GM, Behmand RA, Harik SI. Defective glucose transport across the blood-brain barrier as a cause of persistent hypoglycorrhachia, seizures, and developmental delay. N Engl J Med. 1991; 325:703–709. PMID: 1714544.
Article3. Brockmann K. The expanding phenotype of GLUT1-deficiency syndrome. Brain Dev. 2009; 31:545–552. PMID: 19304421.
Article4. Leen WG, Klepper J, Verbeek MM, Leferink M, Hofste T, van Engelen BG, et al. Glucose transporter-1 deficiency syndrome: the expanding clinical and genetic spectrum of a treatable disorder. Brain. 2010; 133(Pt 3):655–670. PMID: 20129935.
Article5. Pons R, Collins A, Rotstein M, Engelstad K, De Vivo DC. The spectrum of movement disorders in Glut-1 deficiency. Mov Disord. 2010; 25:275–281. PMID: 20063428.
Article6. Leen WG, Willemsen MA, Wevers RA, Verbeek MM. Cerebrospinal fluid glucose and lactate: age-specific reference values and implications for clinical practice. PLoS One. 2012; 7:e42745. PMID: 22880096.
Article7. Hully M, Vuillaumier-Barrot S, Le Bizec C, Boddaert N, Kaminska A, Lascelles K, et al. From splitting GLUT1 deficiency syndromes to overlapping phenotypes. Eur J Med Genet. 2015; 58:443–454. PMID: 26193382.
Article8. Alter AS, Engelstad K, Hinton VJ, Montes J, Pearson TS, Akman CI, et al. Long-term clinical course of Glut1 deficiency syndrome. J Child Neurol. 2015; 30:160–169. PMID: 24789115.
Article9. Lebon S, Suarez P, Alija S, Korff CM, Fluss J, Mercati D, et al. When should clinicians search for GLUT1 deficiency syndrome in childhood generalized epilepsies? Eur J Paediatr Neurol. 2015; 19:170–175. PMID: 25532859.
Article10. Ragona F, Matricardi S, Castellotti B, Patrini M, Freri E, Binelli S, et al. Refractory absence epilepsy and glut1 deficiency syndrome: a new case report and literature review. Neuropediatrics. 2014; 45:328–332. PMID: 24892788.
Article11. Gramer G, Wolf NI, Vater D, Bast T, Santer R, Kamsteeg EJ, et al. Glucose transporter-1 (GLUT1) deficiency syndrome: diagnosis and treatment in late childhood. Neuropediatrics. 2012; 43:168–171. PMID: 22622956.
Article12. De Giorgis V, Varesio C, Baldassari C, Piazza E, Olivotto S, Macasaet J, et al. Atypical manifestations in Glut1 deficiency syndrome. J Child Neurol. 2016; 31:1174–1180. PMID: 27250207.
Article13. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015; 17:405–424. PMID: 25741868.
Article14. Leen WG, Taher M, Verbeek MM, Kamsteeg EJ, van de Warrenburg BP, Willemsen MA. GLUT1 deficiency syndrome into adulthood: a follow-up study. J Neurol. 2014; 261:589–599. PMID: 24413642.
Article15. Chugani HT. A critical period of brain development: studies of cerebral glucose utilization with PET. Prev Med. 1998; 27:184–188. PMID: 9578992.
Article16. Veggiotti P, De Giorgis V. Dietary treatments and new therapeutic perspective in GLUT1 deficiency syndrome. Curr Treat Options Neurol. 2014; 16:291. PMID: 24634059.
Article17. Leen WG, Mewasingh L, Verbeek MM, Kamsteeg EJ, van de Warrenburg BP, Willemsen MA. Movement disorders in GLUT1 deficiency syndrome respond to the modified Atkins diet. Mov Disord. 2013; 28:1439–1442. PMID: 23801573.
Article18. Klepper J, Scheffer H, Leiendecker B, Gertsen E, Binder S, Leferink M, et al. Seizure control and acceptance of the ketogenic diet in GLUT1 deficiency syndrome: a 2-to 5-year follow-up of 15 children enrolled prospectively. Neuropediatrics. 2005; 36:302–308. PMID: 16217704.19. De Giorgis V, Teutonico F, Cereda C, Balottin U, Bianchi M, Giordano L, et al. Sporadic and familial glut1ds Italian patients: a wide clinical variability. Seizure. 2015; 24:28–32. PMID: 25564316.
Article20. Oguni H, Ito Y, Otani Y, Nagata S. Questionnaire survey on the current status of ketogenic diet therapy in patients with glucose transporter 1 deficiency syndrome (GLUT1DS) in Japan. Eur J Paediatr Neurol. 2018; 22:482–487. PMID: 29307699.
Article21. Kass HR, Winesett SP, Bessone SK, Turner Z, Kossoff EH. Use of dietary therapies amongst patients with GLUT1 deficiency syndrome. Seizure. 2016; 35:83–87. PMID: 26803281.
Article22. Fujii T, Ito Y, Takahashi S, Shimono K, Natsume J, Yanagihara K, et al. Outcome of ketogenic diets in GLUT1 deficiency syndrome in Japan: a nationwide survey. Brain Dev. 2016; 38:628–637. PMID: 26923720.
Article23. Anheim M, Maillart E, Vuillaumier-Barrot S, Flamand-Rouvière C, Pineau F, Ewenczyk C, et al. Excellent response to acetazolamide in a case of paroxysmal dyskinesias due to GLUT1-deficiency. J Neurol. 2011; 258:316–317. PMID: 20830593.
Article24. Di Vito L, Licchetta L, Pippucci T, Baldassari S, Stipa C, Mostacci B, et al. Phenotype variability of GLUT1 deficiency syndrome: Description of a case series with novel SLC2A1 gene mutations. Epilepsy Behav. 2018; 79:169–173. PMID: 29306089.
Article25. Barca E, Tang M, Kleiner G, Engelstad K, DiMauro S, Quinzii CM, et al. CoQ10 deficiency is not a common finding in GLUT1 deficiency syndrome. JIMD Rep. 2016; 29:47–52. PMID: 26615598.
Article26. Wang D, Kranz-Eble P, De Vivo DC. Mutational analysis of GLUT1 (SLC2A1) in Glut-1 deficiency syndrome. Hum Mutat. 2000; 16:224–231. PMID: 10980529.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Evolving Diagnostic Criteria in Amyotrophic Lateral Sclerosis and Its Differential Diagnosis
- GLUT1 as a Prognostic Factor for Classical Hodgkin's Lymphoma: Correlation with PD-L1 and PD-L2 Expression
- Prognosis of GLUT1 Expression in Human Breast Carcinoma
- Prognosis of GLUT1 Expression in Human Breast Carcinoma
- Glucose transporter 1 (GLUT1) expression is associated with intestinal type of gastric carcinoma
