Diagnostic approaches for inherited hemolytic anemia in the genetic era
- Affiliations
-
- 1Department of Laboratory Medicine, Catholic Genetic Laboratory Center, Seoul St. Mary's Hospital, College of Medicine, The Catholic University of Korea, Seoul, Korea. microkim@catholic.ac.kr
- KMID: 2413324
- DOI: http://doi.org/10.5045/br.2017.52.2.84
Abstract
- Inherited hemolytic anemias (IHAs) are genetic diseases that present with anemia due to the increased destruction of circulating abnormal RBCs. The RBC abnormalities are classified into the three major disorders of membranopathies, hemoglobinopathies, and enzymopathies. Traditional diagnosis of IHA has been performed via a step-wise process combining clinical and laboratory findings. Nowadays, the etiology of IHA accounts for germline mutations of the responsible genes coding for the structural components of RBCs. Recent advances in molecular technologies, including next-generation sequencing, inspire us to apply these technologies as a first-line approach for the identification of potential mutations and to determine the novel causative genes in patients with IHAs. We herein review the concept and strategy for the genetic diagnosis of IHAs and provide an overview of the preparations for clinical applications of the new molecular technologies.
MeSH Terms
Figure
-
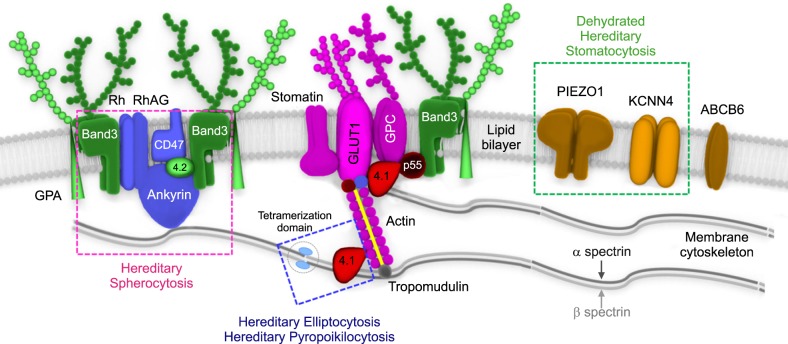
Fig. 1 A schematic representation of red blood cell (RBC) membrane structure with major functional components. The RBC membrane consists of three basic components: a lipid bilayer, transmembrane proteins, and a cytoskeletal network. The major transmembrane proteins are glycoproteins, band 3, and glycophorin. The most abundant protein in the membrane skeleton is spectrin, which is tethered to the phospholipid membrane.Abbreviations: 4.1, protein band 4.1; 4.2, protein band 4.2; GLUT1, glucose transporter 1; GPA, glycophorin A; GPC, glycophorin C; Rh, rhesus polypeptide; RhAG, Rh-associated glycoprotein.
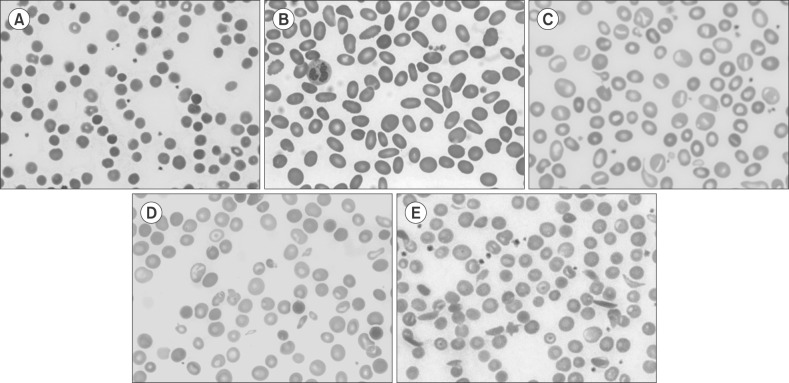
Fig. 2 Peripheral blood smear of inherited hemolytic anemia. (A) Hereditary spherocytosis, (B) hereditary elliptocytosis, (C) hereditary stomatocytosis, (D) β-thalassemia, (E) sickle cell anemia.
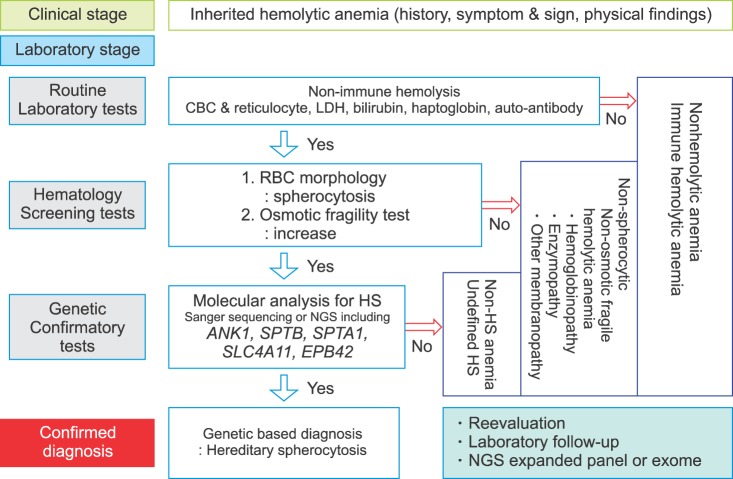
Fig. 3 Stepwise process for genetic-based diagnosis of hereditary spherocytosis.Abbreviations: CBC, complete blood cell counting; HS, hereditary spherocytosis; LDH, lactate dehydrogenase; NGS, next-generation sequencing; RBC, red blood cell.
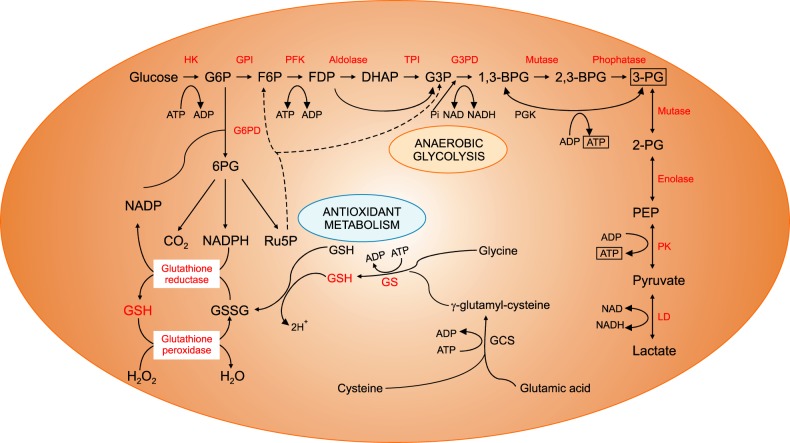
Fig. 4 Anaerobic glycolysis and antioxidant metabolic pathways of red blood cells.Abbreviations: BPG, bisphosphoglyceric acid; DHAP, dihydroxyacetone phosphate; F6P, fructose 6-phosphate; FDP, fructose-1,6-diphosphate; G3P, glycerol 3-phosphate; G3PD, glyceraldehyde 3-phosphate dehydrogenase; G6P, glucose 6-phosphate; G6PD, glucose-6-phosphate dehydrogenase; GCS, glutamylcysteine synthetase; GPI, glucose-phosphate isomerase; GS, glutathione synthetase; GSH, glutathione; GSSG, glutathione disulfide; HK, hexokinase; LD, lactate dehydrogenase; NADP, nicotinamide adenine dinucleotide phosphate; PEP, phosphoenolpyruvic acid; PFK, phosphofructokinase; PG, phosphoglyceric acid; PGK, phosphoglycerate kinase; PK, pyruvate kinase; Ru5P, ribose-5-phosphate isomerase.
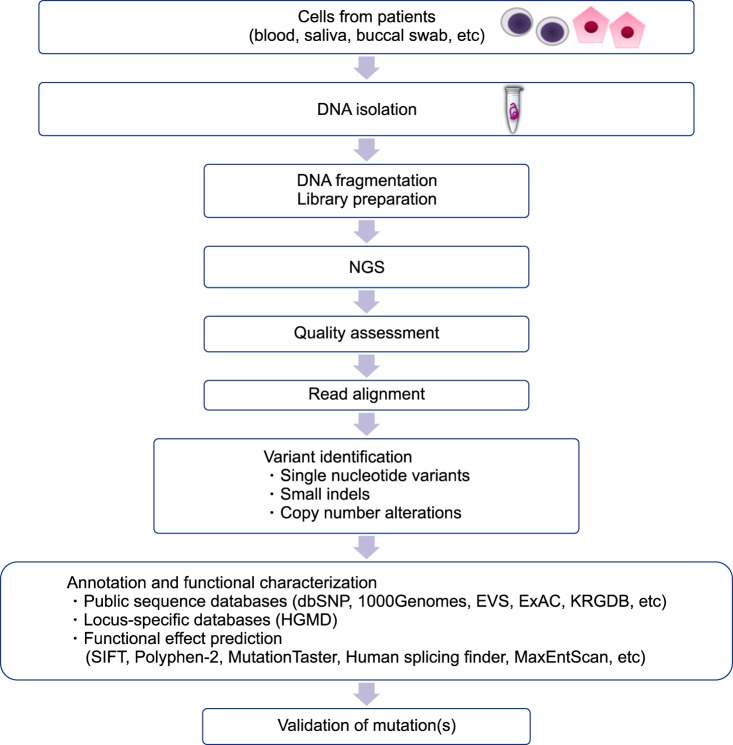
Fig. 5 Overview of steps in the generation of NGS data and analysis.Abbreviations: NGS, next-generation sequencing; dbSNP, NCBI dbSNP Build 141, http://www.ncbi.nlm.nih.gov/projects/SNP/; 1000Genomes, 1000 Genomes Project, http://www.1000genomes.org/; EVS, Exome Variant Server, http://evs.gs.washington.edu/EVS/; ExAC, Exome Aggregation Consortium database, http://exac.broadinstitute.org/; KRDGB, Korean Reference Genome Database, http://152.99.75.168/KRGDB/menuPages/intro.jsp; SIFT, http://sift.jcvi.org/; PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/; MutationTaster, http://www.mutationtaster.org/; Human splicing findinger, http://www.umd.be/HSF/; MaxEntScan, http://genes.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html.
Reference
-
1. Haley K. Congenital hemolytic anemia. Med Clin North Am. 2017; 101:361–374. PMID: 28189176.
Article2. Ucar K. Clinical presentation and management of hemolytic anemias. Oncology (Williston Park). 2002; 16(9 Suppl 10):163–170. PMID: 12380967.3. Lode HN, Krings G, Schulze-Neick I, et al. Pulmonary hypertension in a case of Hb-Mainz hemolytic anemia. J Pediatr Hematol Oncol. 2007; 29:173–177. PMID: 17356397.
Article4. Chiu D, Lubin B. Oxidative hemoglobin denaturation and RBC destruction: the effect of heme on red cell membranes. Semin Hematol. 1989; 26:128–135. PMID: 2658088.5. Jacobasch G, Rapoport SM. Hemolytic anemias due to erythrocyte enzyme deficiencies. Mol Aspects Med. 1996; 17:143–170. PMID: 8813716.6. Zanella A, Fermo E, Bianchi P, Valentini G. Red cell pyruvate kinase deficiency: molecular and clinical aspects. Br J Haematol. 2005; 130:11–25. PMID: 15982340.
Article7. Parker CJ. Paroxysmal nocturnal hemoglobinuria. Curr Opin Hematol. 2012; 19:141–148. PMID: 22395662.
Article8. Gallagher PG. Diagnosis and management of rare congenital nonimmune hemolytic disease. Hematology Am Soc Hematol Educ Program. 2015; 2015:392–399. PMID: 26637748.
Article9. Park ES, Jung HL, Kim HJ, et al. Hereditary hemolytic anemia in Korea from 2007 to 2011: A study by the Korean Hereditary Hemolytic Anemia Working Party of the Korean Society of Hematology. Blood Res. 2013; 48:211–216. PMID: 24086942.
Article10. Barcellini W, Fattizzo B. Clinical applications of hemolytic markers in the differential diagnosis and management of hemolytic anemia. Dis Markers. 2015; 2015:635670. PMID: 26819490.
Article11. King MJ, Zanella A. Hereditary red cell membrane disorders and laboratory diagnostic testing. Int J Lab Hematol. 2013; 35:237–243. PMID: 23480868.
Article12. Christensen RD, Nussenzveig RH, Yaish HM, Henry E, Eggert LD, Agarwal AM. Causes of hemolysis in neonates with extreme hyperbilirubinemia. J Perinatol. 2014; 34:616–619. PMID: 24762414.
Article13. van der Harst P, Zhang W, Mateo Leach I, et al. Seventy-five genetic loci influencing the human red blood cell. Nature. 2012; 492:369–375. PMID: 23222517.14. Sankaran VG, Gallagher PG. Applications of high-throughput DNA sequencing to benign hematology. Blood. 2013; 122:3575–3582. PMID: 24021670.
Article15. Agarwal AM, Nussenzveig RH, Reading NS, et al. Clinical utility of next-generation sequencing in the diagnosis of hereditary haemolytic anaemias. Br J Haematol. 2016; 174:806–814. PMID: 27292444.
Article16. Andolfo I, Russo R, Gambale A, Iolascon A. New insights on hereditary erythrocyte membrane defects. Haematologica. 2016; 101:1284–1294. PMID: 27756835.
Article17. Shohet SB, Bicknese SE. Defining the architecture of the red blood cell membrane: newer biophysical approaches. Am J Hematol. 1993; 42:19–24. PMID: 8416291.
Article18. Kusumi A, Sako Y. Cell surface organization by the membrane skeleton. Curr Opin Cell Biol. 1996; 8:566–574. PMID: 8791449.
Article19. Mohandas N, Gallagher PG. Red cell membrane: past, present, and future. Blood. 2008; 112:3939–3948. PMID: 18988878.
Article20. Palek J. Introduction: red blood cell membrane proteins, their genes and mutations. Semin Hematol. 1993; 30:1–3.21. An X, Mohandas N. Disorders of red cell membrane. Br J Haematol. 2008; 141:367–375. PMID: 18341630.
Article22. Gallagher PG. Update on the clinical spectrum and genetics of red blood cell membrane disorders. Curr Hematol Rep. 2004; 3:85–91. PMID: 14965483.23. Bolton-Maggs PH, Langer JC, Iolascon A, Tittensor P, King MJ. General Haematology Task Force of the British Committee for Standards in Haematology. Guidelines for the diagnosis and management of hereditary spherocytosis-2011 update. Br J Haematol. 2012; 156:37–49. PMID: 22055020.24. Bianchi P, Fermo E, Vercellati C, et al. Diagnostic power of laboratory tests for hereditary spherocytosis: a comparison study in 150 patients grouped according to molecular and clinical characteristics. Haematologica. 2012; 97:516–523. PMID: 22058213.
Article25. King MJ, Garçon L, Hoyer JD, et al. ICSH guidelines for the laboratory diagnosis of nonimmune hereditary red cell membrane disorders. Int J Lab Hematol. 2015; 37:304–325. PMID: 25790109.
Article26. King MJ, Behrens J, Rogers C, Flynn C, Greenwood D, Chambers K. Rapid flow cytometric test for the diagnosis of membrane cytoskeleton-associated haemolytic anaemia. Br J Haematol. 2000; 111:924–933. PMID: 11122157.
Article27. Farias MG. Advances in laboratory diagnosis of hereditary spherocytosis. Clin Chem Lab Med. 2017; 55:944–948. PMID: 27837594.
Article28. Frickhofen N, Chen ZJ, Young NS, Cohen BJ, Heimpel H, Abkowitz JL. Parvovirus B19 as a cause of acquired chronic pure red cell aplasia. Br J Haematol. 1994; 87:818–824. PMID: 7986722.
Article29. Hassoun H, Vassiliadis JN, Murray J, et al. Characterization of the underlying molecular defect in hereditary spherocytosis associated with spectrin deficiency. Blood. 1997; 90:398–406. PMID: 9207476.30. Mariani M, Barcellini W, Vercellati C, et al. Clinical and hematologic features of 300 patients affected by hereditary spherocytosis grouped according to the type of the membrane protein defect. Haematologica. 2008; 93:1310–1317. PMID: 18641031.
Article31. Delaunay J. The molecular basis of hereditary red cell membrane disorders. Blood Rev. 2007; 21:1–20. PMID: 16730867.
Article32. Iolascon A, Miraglia del Giudice E, Perrotta S, Alloisio N, Morlé L, Delaunay J. Hereditary spherocytosis: from clinical to molecular defects. Haematologica. 1998; 83:240–257. PMID: 9573679.33. Eber SW, Gonzalez JM, Lux ML, et al. Ankyrin-1 mutations are a major cause of dominant and recessive hereditary spherocytosis. Nat Genet. 1996; 13:214–218. PMID: 8640229.
Article34. Eber S, Lux SE. Hereditary spherocytosis--defects in proteins that connect the membrane skeleton to the lipid bilayer. Semin Hematol. 2004; 41:118–141. PMID: 15071790.
Article35. Park J, Jeong DC, Yoo J, et al. Mutational characteristics of ANK1 and SPTB genes in hereditary spherocytosis. Clin Genet. 2016; 90:69–78. PMID: 26830532.36. Da Costa L, Galimand J, Fenneteau O, Mohandas N. Hereditary spherocytosis, elliptocytosis, and other red cell membrane disorders. Blood Rev. 2013; 27:167–178. PMID: 23664421.
Article37. Glele-Kakai C, Garbarz M, Lecomte MC, et al. Epidemiological studies of spectrin mutations related to hereditary elliptocytosis and spectrin polymorphisms in Benin. Br J Haematol. 1996; 95:57–66. PMID: 8857939.38. Dhermy D, Schrével J, Lecomte MC. Spectrin-based skeleton in red blood cells and malaria. Curr Opin Hematol. 2007; 14:198–202. PMID: 17414207.
Article39. Gallagher PG, Weed SA, Tse WT, et al. Recurrent fatal hydrops fetalis associated with a nucleotide substitution in the erythrocyte beta-spectrin gene. J Clin Invest. 1995; 95:1174–1182. PMID: 7883966.
Article40. Cho HS, Hah JO, Kang IJ, et al. Hereditary hemolytic anemia in Korea: a retrospective study from 1997 to 2006. Korean J Hematol. 2007; 42:197–205.
Article41. Han E, Kim A, Park J, et al. Spectrin Tunis (Sp alpha (I/78)) in a Korean family with hereditary elliptocytosis. Ann Lab Med. 2013; 33:386–389. PMID: 24003435.
Article42. Hoffman R, Benz EJ Jr, Silberstein LE, Heslop H, Weitz J, Anastasi J. Hematology: basic principles and practice. 6th ed. PA: Elsevier Saunders;2013. p. 418–426.43. Niss O, Chonat S, Dagaonkar N, et al. Genotype-phenotype correlations in hereditary elliptocytosis and hereditary pyropoikilocytosis. Blood Cells Mol Dis. 2016; 61:4–9. PMID: 27667160.
Article44. Prchal JT, Gregg XT. Red cell enzymes. Hematology Am Soc Hematol Educ Program. 2005; 19–23. PMID: 16304354.
Article45. Koralkova P, van Solinge WW, van Wijk R. Rare hereditary red blood cell enzymopathies associated with hemolytic anemia - pathophysiology, clinical aspects, and laboratory diagnosis. Int J Lab Hematol. 2014; 36:388–397. PMID: 24750686.
Article46. Vives i Corrons JL. Chronic non-spherocytic haemolytic anaemia due to congenital pyrimidine 5' nucleotidase deficiency: 25 years later. Baillieres Best Pract Res Clin Haematol. 2000; 13:103–118. PMID: 10916681.47. Kahn A, Kaplan JC, Dreyfus JC. Advances in hereditary red cell enzyme anomalies. Hum Genet. 1979; 50:1–27. PMID: 157322.
Article48. Fiorelli G, Martinez di Montemuros F, Cappellini MD. Chronic non-spherocytic haemolytic disorders associated with glucose-6-phosphate dehydrogenase variants. Baillieres Best Pract Res Clin Haematol. 2000; 13:39–55. PMID: 10916677.
Article49. Zanella A, Fermo E, Bianchi P, Chiarelli LR, Valentini G. Pyruvate kinase deficiency: the genotype-phenotype association. Blood Rev. 2007; 21:217–231. PMID: 17360088.
Article50. Canu G, De Bonis M, Minucci A, Capoluongo E. Red blood cell PK deficiency: An update of PK-LR gene mutation database. Blood Cells Mol Dis. 2016; 57:100–109. PMID: 26832193.
Article51. Cappellini MD, Fiorelli G. Glucose-6-phosphate dehydrogenase deficiency. Lancet. 2008; 371:64–74. PMID: 18177777.
Article52. Howes RE, Battle KE, Satyagraha AW, Baird JK, Hay SI. G6PD deficiency: global distribution, genetic variants and primaquine therapy. Adv Parasitol. 2013; 81:133–201. PMID: 23384623.53. von Seidlein L, Auburn S, Espino F, et al. Review of key knowledge gaps in glucose-6-phosphate dehydrogenase deficiency detection with regard to the safe clinical deployment of 8-aminoquinoline treatment regimens: a workshop report. Malar J. 2013; 12:112. PMID: 23537118.
Article54. Keihanian F, Basirjafari S, Darbandi B, et al. Comparison of quantitative and qualitative tests for glucose-6-phosphate dehydrogenase deficiency in the neonatal period. Int J Lab Hematol. 2017; 39:251–260. PMID: 28258653.
Article55. Nadarajan V, Shanmugam H, Sthaneshwar P, et al. Modification to reporting of qualitative fluorescent spot test results improves detection of glucose-6-phosphate dehydrogenase (G6PD)-deficient heterozygote female newborns. Int J Lab Hematol. 2011; 33:463–470. PMID: 21501392.
Article56. Minucci A, Giardina B, Zuppi C, et al. Glucose-6-phosphate dehydrogenase laboratory assay: How, when, and why? IUBMB Life. 2009; 61:27–34. PMID: 18942156.
Article57. Glucose-6-phosphate dehydrogenase deficiency. WHO Working Group. Bull World Health Organ. 1989; 67:601–611. PMID: 2633878.58. Vaca G, Arámbula E, Monsalvo A, et al. Glucose-6-phosphate dehydrogenase (G-6-PD) mutations in Mexico: four new G-6-PD variants. Blood Cells Mol Dis. 2003; 31:112–120. PMID: 12850494.
Article59. Domingo GJ, Satyagraha AW, Anvikar A, et al. G6PD testing in support of treatment and elimination of malaria: recommendations for evaluation of G6PD tests. Malar J. 2013; 12:391. PMID: 24188096.
Article60. Minucci A, Moradkhani K, Hwang MJ, Zuppi C, Giardina B, Capoluongo E. Glucose-6-phosphate dehydrogenase (G6PD) mutations database: review of the "old" and update of the new mutations. Blood Cells Mol Dis. 2012; 48:154–165. PMID: 22293322.
Article61. Lee J, Park J, Choi H, et al. Genetic profiles of Korean patients with glucose-6-phosphate dehydrogenase deficiency. Ann Lab Med. 2017; 37:108–116. PMID: 28028996.
Article62. Gómez-Manzo S, Marcial-Quino J, Vanoye-Carlo A, et al. Glucose-6-phosphate dehydrogenase: update and analysis of new mutations around the world. Int J Mol Sci. 2016; 17:E2069. PMID: 27941691.
Article63. Luzzatto L, Nannelli C, Notaro R. Glucose-6-phosphate dehydrogenase deficiency. Hematol Oncol Clin North Am. 2016; 30:373–393. PMID: 27040960.
Article64. Bogari NM. Next generation sequencing (NGS) in glucose-6-phosphate dehydrogenase (G6PD) deficiency studies. Bioinformation. 2016; 12:41–43. PMID: 28104958.
Article65. Beutler E, Baronciani L. Mutations in pyruvate kinase. Hum Mutat. 1996; 7:1–6. PMID: 8664896.
Article66. Climent F, Roset F, Repiso A, Pérez de la Ossa P. Red cell glycolytic enzyme disorders caused by mutations: an update. Cardiovasc Hematol Disord Drug Targets. 2009; 9:95–106. PMID: 19519368.
Article67. Grace RF, Zanella A, Neufeld EJ, et al. Erythrocyte pyruvate kinase deficiency: 2015 status report. Am J Hematol. 2015; 90:825–830. PMID: 26087744.
Article68. Rider NL, Strauss KA, Brown K, et al. Erythrocyte pyruvate kinase deficiency in an old-order Amish cohort: longitudinal risk and disease management. Am J Hematol. 2011; 86:827–834. PMID: 21815188.
Article69. Kim M, Park J, Lee J, et al. Hemolytic anemia with null PKLR mutations identified using whole exome sequencing and cured by hematopoietic stem cell transplantation combined with splenectomy. Bone Marrow Transplant. 2016; 51:1605–1608. PMID: 27595284.
Article70. Weatherall DJ. Hemoglobinopathies worldwide: present and future. Curr Mol Med. 2008; 8:592–599. PMID: 18991645.
Article71. Wang HC, Hsieh LL, Liu YC, et al. The epidemiologic transition of thalassemia and associated hemoglobinopathies in southern Taiwan. Ann Hematol. 2017; 96:183–188. PMID: 27891555.
Article72. Higgs DR, Weatherall DJ. The alpha thalassaemias. Cell Mol Life Sci. 2009; 66:1154–1162. PMID: 19020805.
Article73. Olivieri NF. The beta-thalassemias. N Engl J Med. 1999; 341:99–109. PMID: 10395635.74. Clarke GM, Higgins TN. Laboratory investigation of hemoglobinopathies and thalassemias: review and update. Clin Chem. 2000; 46:1284–1290. PMID: 10926923.
Article75. Kutlar F. Diagnostic approach to hemoglobinopathies. Hemoglobin. 2007; 31:243–250. PMID: 17486507.
Article76. Patrinos GP, Kollia P, Papadakis MN. Molecular diagnosis of inherited disorders: lessons from hemoglobinopathies. Hum Mutat. 2005; 26:399–412. PMID: 16138310.
Article77. Colosimo A, Gatta V, Guida V, et al. Application of MLPA assay to characterize unsolved a-globin gene rearrangements. Blood Cells Mol Dis. 2011; 46:139–144. PMID: 21190870.78. Kipp BR, Roellinger SE, Lundquist PA, Highsmith WE, Dawson DB. Development and clinical implementation of a combination deletion PCR and multiplex ligation-dependent probe amplification assay for detecting deletions involving the human a-globin gene cluster. J Mol Diagn. 2011; 13:549–557. PMID: 21708285.79. Higgins T. Hemoglobinopathies/thalassemias: Why clinical biochemists need to know about them. Clin Biochem. 2017; [Epub ahead of print].
Article80. Brennan SO. Fifty-eight years of hemoglobin analysis. Clin Chem. 2008; 54:8–10. PMID: 18160724.
Article81. Giardine B, Borg J, Viennas E, et al. Updates of the HbVar database of human hemoglobin variants and thalassemia mutations. Nucleic Acids Res. 2014; 42:D1063–D1069. PMID: 24137000.
Article82. Jo I, Jang W, Chae H, et al. Hemoglobin Kansas: first Korean family and literature review. Ann Lab Med. 2017; 37:352–354. PMID: 28445020.
Article83. Lee ST, Kim MS, Choi DY, Kim SK, Ki CS. Incidence of variant hemoglobin (Hb) and increased fetal Hb concentrations and their effect on Hb A1c measurement in a Korean population. Clin Chem. 2006; 52:1445–1446. PMID: 16798979.
Article84. Jung CL, Kwon KJ, Hong KS, et al. Hemoglobin Yamagata: hemoglobin variant detected by HbA1c test. Korean J Lab Med. 2009; 29:536–540. PMID: 20046085.
Article85. He J, Song W, Yang J, et al. Next-generation sequencing improves thalassemia carrier screening among premarital adults in a high prevalence population: the Dai nationality, China. Genet Med. 2017; [Epub ahead of print].
Article86. Goodman MA, Malik P. The potential of gene therapy approaches for the treatment of hemoglobinopathies: achievements and challenges. Ther Adv Hematol. 2016; 7:302–315. PMID: 27695619.
Article87. Traxler EA, Yao Y, Wang YD, et al. A genome-editing strategy to treat β-hemoglobinopathies that recapitulates a mutation associated with a benign genetic condition. Nat Med. 2016; 22:987–990. PMID: 27525524.
Article88. Rai P, Malik P. Gene therapy for hemoglobin disorders - a mini-review. J Rare Dis Res Treat. 2016; 1:25–31. PMID: 27891535.
Article89. Jamwal M, Aggarwal A, Das A, et al. Next-generation sequencing unravels homozygous mutation in glucose-6-phosphate isomerase, GPIc.1040G>A (p.Arg347His) causing hemolysis in an Indian infant. Clin Chim Acta. 2017; 468:81–84. PMID: 28223188.90. Del Orbe Barreto R, Arrizabalaga B, De la Hoz AB, et al. Detection of new pathogenic mutations in patients with congenital haemolytic anaemia using next-generation sequencing. Int J Lab Hematol. 2016; 38:629–638. PMID: 27427187.
Article91. Xue Y, Ankala A, Wilcox WR, Hegde MR. Solving the molecular diagnostic testing conundrum for Mendelian disorders in the era of next-generation sequencing: single-gene, gene panel, or exome/genome sequencing. Genet Med. 2015; 17:444–451. PMID: 25232854.
Article92. Chrystoja CC, Diamandis EP. Whole genome sequencing as a diagnostic test: challenges and opportunities. Clin Chem. 2014; 60:724–733. PMID: 24227285.
Article93. Han JH, Kim S, Jang H, et al. Identification of a novel p.Q1772X ANK1 mutation in a Korean family with hereditary spherocytosis. PLoS One. 2015; 10:e0131251. PMID: 26107955.
Article94. Lacy JN, Ulirsch JC, Grace RF, et al. Exome sequencing results in successful diagnosis and treatment of a severe congenital anemia. Cold Spring Harb Mol Case Stud. 2016; 2:a000885. PMID: 27551681.
Article95. Lettre G. The search for genetic modifiers of disease severity in the β-hemoglobinopathies. Cold Spring Harb Perspect Med. 2012; 2:pii:a015032.
Article96. Wooderchak-Donahue WL, O'Fallon B, Furtado LV, et al. A direct comparison of next generation sequencing enrichment methods using an aortopathy gene panel- clinical diagnostics perspective. BMC Med Genomics. 2012; 5:50. PMID: 23148498.
Article97. Dames S, Chou LS, Xiao Y, et al. The development of next-generation sequencing assays for the mitochondrial genome and 108 nuclear genes associated with mitochondrial disorders. J Mol Diagn. 2013; 15:526–534. PMID: 23665194.
Article98. Sun Y, Ruivenkamp CA, Hoffer MJ, et al. Next-generation diagnostics: gene panel, exome, or whole genome? Hum Mutat. 2015; 36:648–655. PMID: 25772376.
Article99. Roy NB, Wilson EA, Henderson S, et al. A novel 33-Gene targeted resequencing panel provides accurate, clinical-grade diagnosis and improves patient management for rare inherited anaemias. Br J Haematol. 2016; 175:318–330. PMID: 27432187.
Article100. Charoenkwan P, Natesirinilkul R, Choeyprasert W, Kulsumritpon N, Sangiamporn O. Coinheritance of hereditary elliptocytosis and deletional hemoglobin H disease. J Pediatr Hematol Oncol. 2017; 39:e69–e70. PMID: 28060122.
Article101. Hoffmann TJ, Witte JS. Strategies for imputing and analyzing rare variants in association studies. Trends Genet. 2015; 31:556–563. PMID: 26450338.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- A new paradigm in the diagnosis of hereditary hemolytic anemia
- A case of Wilson disease associated with hemolytic anemia and cholelithiasis
- Two Cases of Autoimmune Hemolytic Anemia in Ulcerative Colitis
- A 22-month-old Boy with Acute Glomerulonephritis Coexistent with Hemolytic Anemia and Idiopathic Thrombocytopenia
- Two Cases of Autoimmune Hemolytic Anemic
