Pediatr Gastroenterol Hepatol Nutr.
2015 Jun;18(2):138-143. 10.5223/pghn.2015.18.2.138.
Clinical, Biochemical, and Genetic Characterization of Glycogen Storage Type IX in a Child with Asymptomatic Hepatomegaly
- Affiliations
-
- 1Department of Pediatrics, Asan Medical Center Children's Hospital, University of Ulsan College of Medicine, Seoul, Korea. kmkim@amc.seoul.kr
- 2Medical Genetics Center, Asan Medical Center Children's Hospital, University of Ulsan College of Medicine, Seoul, Korea.
- 3University Children's Hospital and Molecular Genetics and Metabolism Laboratory, Munich, Germany.
- KMID: 2315562
- DOI: http://doi.org/10.5223/pghn.2015.18.2.138
Abstract
- Glycogen storage disease type IX (GSD IX) is caused by a defect in phosphorylase b kinase (PhK) that results from mutations in the PHKA2, PHKB, and PHKG2 genes. Patients usually manifest recurrent ketotic hypoglycemia with growth delay, but some may present simple hepatomegaly. Although GSD IX is one of the most common causes of GSDs, its biochemical and genetic diagnosis has been problematic due to its rarity, phenotypic overlap with other types of GSDs, and genetic heterogeneities. In our report, a 22-month-old boy with GSD IX is described. No other manifestations were evident except for hepatomegaly. His growth and development also have been proceeding normally. Diagnosed was made by histologic examination, an enzyme assay, and genetic testing with known c.3210_3212del (p.Arg1070del) mutation in PHKA2 gene.
Keyword
MeSH Terms
Figure
-

Fig. 1 Pathologic findings. (A) Hepatocytes were filled with abundant glycogen particles in the cytoplasmonroutine transmission electron microscopy (×200). (B) Hepatocyte cytoplasmic material was positive byperiodic acid-Schiff staining (×200). (C) Hepatocyte cytoplasmic material was digested by diastase-treatment (×200).
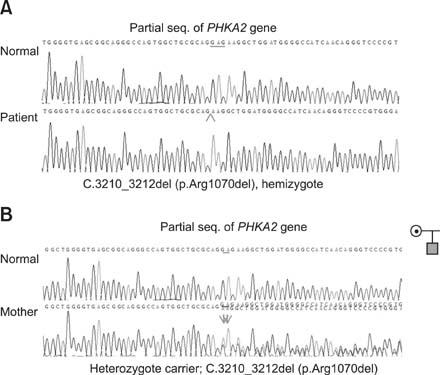
Fig. 2 Partial genomic sequences of the PHAK2 gene. (A) The patient is a homozygotefor the c.3210_3212del (p.Arg1070del) mutation. (B) His mother is a heterozygote for this mutation.
Reference
-
1. Lau CK, Hui J, Fong FN, To KF, Fok TF, Tang NL, et al. Novel mutations in PHKA2 gene in glycogen storage disease type IX patients from Hong Kong, China. Mol Genet Metab. 2011; 102:222–225.
Article2. Maichele AJ, Burwinkel B, Maire I, Søvik O, Kilimann MW. Mutations in the testis/liver isoform of the phosphorylase kinase gamma subunit (PHKG2) cause autosomal liver glycogenosis in the gsd rat and in humans. Nat Genet. 1996; 14:337–340.
Article3. Newgard CB, Hwang PK, Fletterick RJ. The family of glycogen phosphorylases: structure and function. Crit Rev Biochem Mol Biol. 1989; 24:69–99.4. Tsilianidis LA, Fiske LM, Siegel S, Lumpkin C, Hoyt K, Wasserstein M, et al. Aggressive therapy improves cirrhosis in glycogen storage disease type IX. Mol Genet Metab. 2013; 109:179–182.
Article5. Bali DS, Goldstein JL, Fredrickson K, Rehder C, Boney A, Austin S, et al. Variability of disease spectrum in children with liver phosphorylase kinase deficiency caused by mutations in the PHKG2 gene. Mol Genet Metab. 2014; 111:309–313.
Article6. Park KJ, Park HD, Lee SY, Ki CS, Choe YH. A novel PHKA2 gross deletion mutation in a Korean patient with X-linked liver glycogenosis type I. Ann Clin Lab Sci. 2011; 41:197–200.7. Crushell E, Treacy EP, Dawe J, Durkie M, Beauchamp NJ. Glycogen storage disease type III in the Irish population. J Inherit Metab Dis. 2010; 33:Suppl 3. S215–S218.
Article8. Choi J, Ko JM, Kim GH, Yoo HW. Clinical manifestation and effect of corn starch on height growth in Korean patients with glycogen storage disease type Ia. J Korean Soc Pediatr Endocrinol. 2007; 12:35–40.9. Beauchamp NJ, Taybert J, Champion MP, Layet V, Heinz-Erian P, Dalton A, et al. High frequency of missense mutations in glycogen storage disease type VI. J Inherit Metab Dis. 2007; 30:722–734.
Article10. Moses SW, Parvari R. The variable presentations of glycogen storage disease type IV: a review of clinical, enzymatic and molecular studies. Curr Mol Med. 2002; 2:177–188.
Article11. Ban HR, Kim KM, Jang JY, Kim GH, You HW, Kim K, et al. Living donor liver transplantation in a Korean child with glycogen storage disease type IV and a GBE1 mutation. Gut Liver. 2009; 3:60–63.
Article12. Brushia RJ, Walsh DA. Phosphorylase kinase: the complexity of its regulation is reflected in the complexity of its structure. Front Biosci. 1999; 4:D618–D641.
Article13. Rudolfová J, Slovácková R, Trbusek M, Pesková K, St'astná S, Kozák L. Identification of three novel mutations in the PHKA2 gene in Czech patients with X-linked liver glycogenosis. J Inherit Metab Dis. 2001; 24:85–87.
Article14. Hidaka F, Sawada H, Matsuyama M, Nunoi H. A novel mutation of the PHKA2 gene in a patient with X-linked liver glycogenosis type 1. Pediatr Int. 2005; 47:687–690.
Article15. Bak H, Cordato D, Carey WF, Milder D. Adult-onset exercise intolerance due to phosphorylase b kinase deficiency. J Clin Neurosci. 2001; 8:286–287.
Article16. Morava E, Wortmann SB, van Essen HZ, Liebrand van Sambeek R, Wevers R, van Diggelen OP. Biochemical characteristics and increased tetraglucoside excretion in patients with phosphorylase kinase deficiency. J Inherit Metab Dis. 2005; 28:703–706.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- A Case of Glycogen Storage Disease with Hypertrophic Cardiomyopathy
- A case of multiple hepatic adenomas and gout with glycogen storage disease type Ia
- Anesthetic Management for the Patient with Von Gierke`s Diseases
- Juvenile Pompe Disease with CNS Involvement: A Case Report
- A Case of Glycogen Storage Disease Type lb
