Pediatr Gastroenterol Hepatol Nutr.
2016 Mar;19(1):76-81. 10.5223/pghn.2016.19.1.76.
Novel Mutations in the CPT1A Gene Identified in the Patient Presenting Jaundice as the First Manifestation of Carnitine Palmitoyltransferase 1A Deficiency
- Affiliations
-
- 1Department of Pediatrics, Seoul National University College of Medicine, Seoul, Korea. jmko@snu.ac.kr
- KMID: 2160479
- DOI: http://doi.org/10.5223/pghn.2016.19.1.76
Abstract
- Carnitine palmitoyltransferase 1A (CPT1A) is an enzyme functioning in mitochondrial fatty acid oxidation (FAO) of the liver. Patients with CPT1A deficiency have impaired mitochondrial FAO and display hypoketotic hypoglycemia and hepatic encephalopathy as typical manifestations. In this report, we present a case of CPT1A deficiency presenting jaundice as the first manifestation. A 1.9 years old boy showed jaundice and elevated levels of free and total carnitine were observed. From direct sequencing analysis of CPT1A, two novel mutations, c.1163+1G>A and c.1393G>A (p.Gly465Arg), were identified. At the age of 2.2 years, hypoglycemia, tachycardia, and altered mental status developed just after cranioplasty for craniosynostosis. High glucose infusion rate was required for recovery of his vital signs and mentality. Diet rich in high carbohydrate, low fat and inclusion of medium chain triglyceride oil resulted in improvement in cholestatic hepatitis and since then the boy has shown normal growth velocity and developmental milestones to date.
MeSH Terms
Figure
-

Fig. 1 Hepatomegaly and nephromegaly was observed onin ultrasonographic examinationy performed at age 1.9 years.
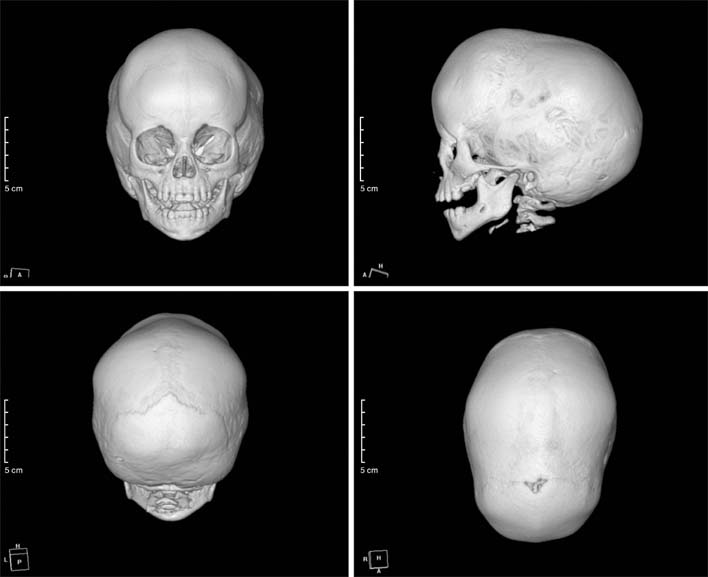
Fig. 2 Three-dimensional reconstruction images of computed tomography show early closure with sclerotic change of the sagittal suture and scaphocephaly.

Fig. 3 Partial genomic DNA sequences of CPT1A for the patient are shown. The patient is a compound heterozygote with two novel mutations, c.1163+1G>A and c.1393G> A (p.Gly465Arg).
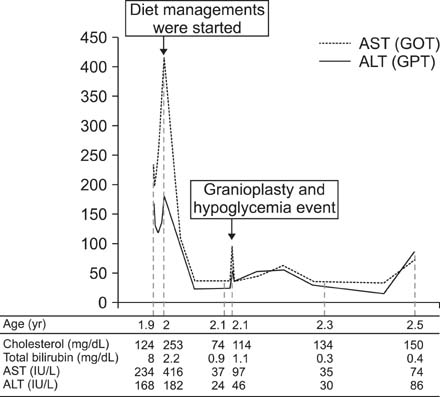
Fig. 4 The course of clinical events and associated laboratory findings is shown as below. AST: aspartate aminotransferase, ALT: alanine transaminase.
Reference
-
1. Collins SA, Sinclair G, McIntosh S, Bamforth F, Thompson R, Sobol I, et al. Carnitine palmitoyltransferase 1A (CPT1A) P479L prevalence in live newborns in Yukon, Northwest Territories, and Nunavut. Mol Genet Metab. 2010; 101:200–204.
Article2. Bennett MJ, Santani AB. Carnitine palmitoyltransferase 1A deficiency. In : Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, editors. GeneReviews(R). Seattle, WA: Seattle University of Washington;1993.3. Lee BH, Kim YM, Kim JH, Kim GH, Kim JM, Kim JH, et al. Atypical manifestation of carnitine palmitoyltransferase 1A deficiency: hepatosplenomegaly and nephromegaly. J Pediatr Gastroenterol Nutr. 2015; 60:e19–e22.4. Bonnefont JP, Djouadi F, Prip-Buus C, Gobin S, Munnich A, Bastin J. Carnitine palmitoyltransferases 1 and 2: biochemical, molecular and medical aspects. Mol Aspects Med. 2004; 25:495–520.
Article5. Morris AA, Olpin SE, Bennett MJ, Santani A, Stahlschmidt J, McClean P. Cholestatic jaundice associated with carnitine palmitoyltransferase IA deficiency. JIMD Rep. 2013; 7:27–29.
Article6. Greenberg CR, Dilling LA, Thompson GR, Seargeant LE, Haworth JC, Phillips S, et al. The paradox of the carnitine palmitoyltransferase type Ia P479L variant in Canadian Aboriginal populations. Mol Genet Metab. 2009; 96:201–207.
Article7. Fingerhut R, Röschinger W, Muntau AC, Dame T, Kreischer J, Arnecke R, et al. Hepatic carnitine palmitoyltransferase I deficiency: acylcarnitine profiles in blood spots are highly specific. Clin Chem. 2001; 47:1763–1768.
Article8. Tsuburaya R, Sakamoto O, Arai N, Kobayashi H, Hasegawa Y, Yamaguchi S, et al. Molecular analysis of a presymptomatic case of carnitine palmitoyl transferase I (CPT I) deficiency detected by tandem mass spectrometry newborn screening in Japan. Brain Dev. 2010; 32:409–411.
Article9. Prasad C, Johnson JP, Bonnefont JP, Dilling LA, Innes AM, Haworth JC, et al. Hepatic carnitine palmitoyl transferase 1 (CPT1 A) deficiency in North American Hutterites (Canadian and American): evidence for a founder effect and results of a pilot study on a DNAbased newborn screening program. Mol Genet Metab. 2001; 73:55–63.
Article10. Brown NF, Mullur RS, Subramanian I, Esser V, Bennett MJ, Saudubray JM, et al. Molecular characterization of L-CPT I deficiency in six patients: insights into function of the native enzyme. J Lipid Res. 2001; 42:1134–1142.
Article11. Bennett MJ, Boriack RL, Narayan S, Rutledge SL, Raff ML. Novel mutations in CPT 1A define molecular heterogeneity of hepatic carnitine palmitoyltransferase I deficiency. Mol Genet Metab. 2004; 82:59–63.
Article12. Falik-Borenstein ZC, Jordan SC, Saudubray JM, Brivet M, Demaugre F, Edmond J, et al. Brief report: renal tubular acidosis in carnitine palmitoyltransferase type 1 deficiency. N Engl J Med. 1992; 327:24–27.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Coexistence of VHL Disease and CPT2 Deficiency: A Case Report
- A Case of Asymptomatic 3-methylcrotonylglycinuria Detected by Tandem Mass Spectrometry in Newborn Screening
- Transient carnitine transport defect with cholestatic jaundice: report of one case in a premature baby
- Primary Carnitine Deficiency and Cardiomyopathy
- A Study for the Normal Serum Carnitine Levels and the Effect of Anticonvulsants on Serum Carnitine Levels in Pediatric Age
