Hypertensive Hypokalemic Disorders
- Affiliations
-
- 1Department of Internal Medicine, College of Medicine, Ewha Womans University, Seoul, Korea. kbchoi@ewha.ac.kr
- KMID: 2134803
- DOI: http://doi.org/10.5049/EBP.2007.5.1.34
Abstract
- Hypokalemia is a common clinical problem. The kidney is responsible for long term potassium homoeostasis, as well as the serum potassium concentration. The main nephron site where K secretion is regulated is the cortical collecting duct, mainly via the effects of aldosterone. Aldosterone interacts with the mineralocorticoid receptor to increase sodium reabsorption and potassium secretion; the removal of cationic sodium makes the lumen relatively electronegative, thereby promoting passive potassium secretion from the tubular cell into the lumen through apical potassium channels. As a result, any condition that decreases the activity of renal potassium channels results in hyperkalemia (for example, amiloride intake or aldosterone deficiency) whereas their increased activity results in hypokalemia (for example, primary aldosteronism or Liddle's syndrome). The cause of hypokalemia can usually be determined from the history. If there is no apparent cause, the initial step is to see if hypokalemia is in associated with systemic hypertension or not. In the former group hypokalaemia is associated with a high mineralocorticoid effect or hyperactive sodium channel as in Liddle's syndrome. In hypertensive hypokalemic patients, measurement of the renin, aldosterone, and cortisol concentrations would be of help in differential diagnosis.
MeSH Terms
-
Aldosterone
Amiloride
Diagnosis, Differential
Humans
Hydrocortisone
Hyperaldosteronism
Hyperkalemia
Hypertension
Hypokalemia
Kidney
Mineralocorticoids
Nephrons
Potassium
Potassium Channels
Receptors, Mineralocorticoid
Renin
Sodium
Sodium Channels
Aldosterone
Amiloride
Hydrocortisone
Mineralocorticoids
Potassium
Potassium Channels
Receptors, Mineralocorticoid
Renin
Sodium
Sodium Channels
Figure
-
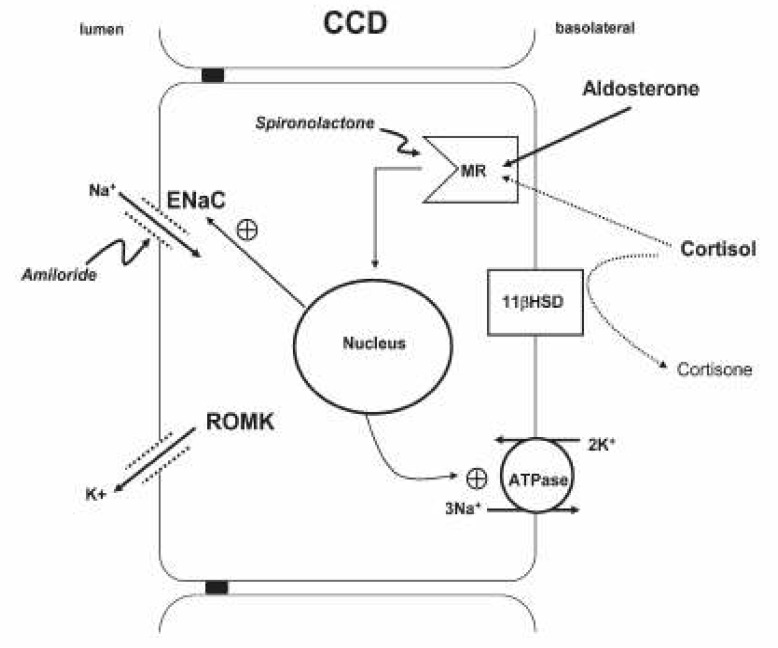
Fig. 1 K+ transport at the principal cell of the cortical collecting duct (CCD). MR, mineralocorticoid receptor; 11β-HSD, 11β-hydroxysteroid dehydrogenase; ENaC, epithelial Na channel; ROMK, luminal ATP-regulated inwardly rectifying K channel. [From Landau D : Cell Mol Life Sci 63:1962-1968, 2006]
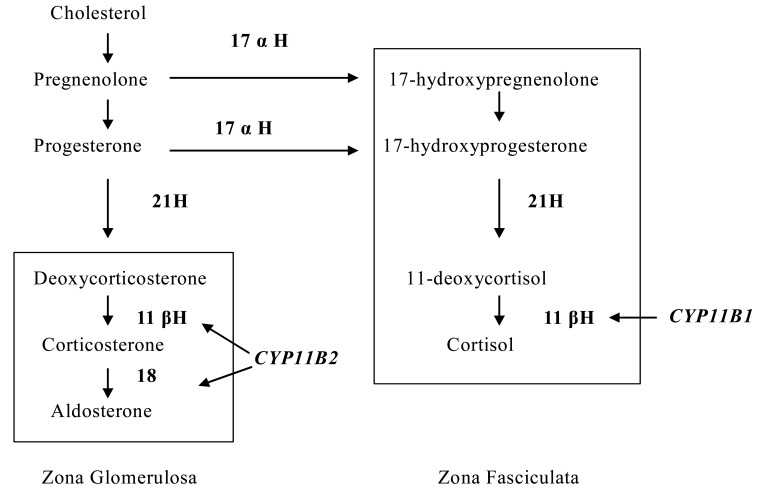
Fig. 2 Normal biosynthetic pathways for cortisol and aldosterone. 17α H, 17α-hydroxylase; 21H, 21-hydroxylase. 11βH, 11-β hydroxylase; 18, 18-hydroxylase/oxidase. [From Freel EM, Connel JNC : J Am Soc Nephrol 15:1993-2001, 2004]
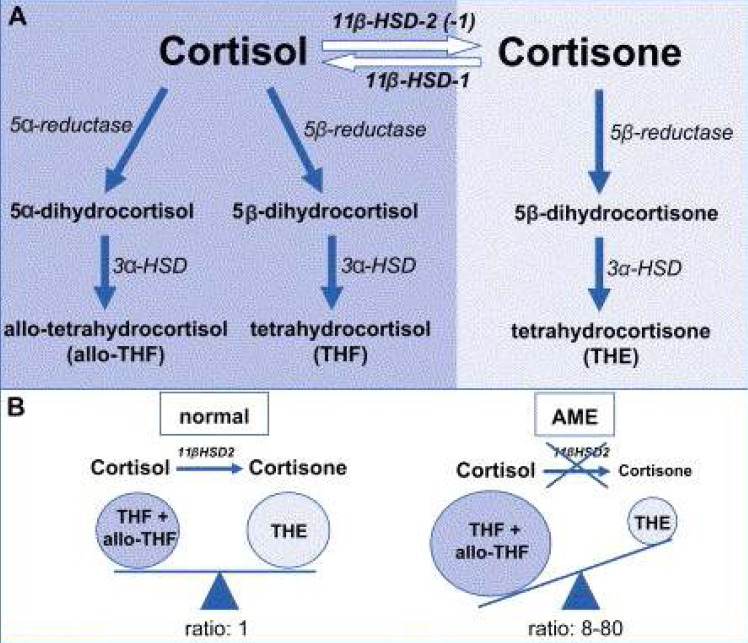
Fig. 3 A) Schematic depiction of the enzymatic activity involved in glucocorticoid metabolism. In the liver, 5α- and 5β-reductases and 3α-hydroxysteroid dehydrogenases (3α-HSDs) convert cortisol to 5α-tetrahydrocortisol (allo-THF) and 5β-tetrahydrocortisol (THF) and convert cortisone to tetrahydrocortisone (THE). B) In normal subjects, urinary excretion of cortisol metabolites compared to the cortisone metabolite is equivalent, resulting in a [THF+allo-THF]/THE ratio of 1. Mutations that inactivate 11β-HSD2 in apparentmineralocorticoid excess (AME) patients result in a grossly increased urinary excretion of THF and allo-THF compounds, whereas THE is dramatically reduced, resulting in a high [THF+allo-THF]/THE ratio. [From Hammer F, Stewart PM : Best Pract Res Clin Endocrinol Metab 20:337-353, 2006]
Reference
-
1. Landau D. Potassium-related inherited tubulopathies. Cell Mol Life Sci. 2006; 63:1962–1968. PMID: 16810456.
Article2. Rastegar A, Soleimani M. Hypokalaemia and hyperkalaemia. Postgrad Med J. 2001; 77:759–764. PMID: 11723313.
Article3. Freel EM, Connell JMR. Mechanisms of hypertension: the expanding role of aldosterone. J Am Soc Nephrol. 2004; 15:1993–2001. PMID: 15284285.
Article4. Hammer F, Stewart PM. Cortisol metabolism in hypertension. Best Pract Res Clin Endocrinol Metab. 2006; 20:337–353. PMID: 16980198.
Article5. Khosla N, Hogan D. Mineralocorticoid hypertension and hypokalemia. Semin Nephrol. 2006; 26:434–440. PMID: 17275580.
Article6. Mattsson C, Young WF Jr. Primary aldosteronism: diagnostic and treatment strategies. Nat Clin Pract Nephrol. 2006; 2:198–208. PMID: 16932426.
Article7. Agharazii M, Douville P, Grose JH, Lebel M. Captopril suppression versus salt loading in confirming primary aldosteronism. Hypertension. 2001; 37:1440–1443. PMID: 11408392.
Article8. McMahon GT, Dluhy RG. Glucocorticoid-remediable aldosteronism. Arq Bras Endocrinol Metabol. 2004; 48:682–686. PMID: 15761539.
Article9. Dluhy RG, Lifton RP. Glucocorticoid-remediable aldosteronism (GRA): diagnosis, variability of phenotype and regulation of potassium homeostasis. Steroids. 1995; 60:48–51. PMID: 7792815.
Article10. Litchfield WR, New MI, Coolidge C, Lifton RP, Dluhy RG. Evaluation of the dexamethasone suppression test for the diagnosis of glucocorticoid-remediable aldosteronism. J Clin Endocrinol Metab. 1997; 82:3570–3573. PMID: 9360508.
Article11. Mosso L, Gomez-Sanchez CE, Foecking MF, Fardella C. Serum 18-hydroxycortisol in primary aldosteronism, hypertension, and normotensives. Hypertension. 2001; 38:688–691. PMID: 11566957.
Article12. Wilson RC, Nimkarn S, New MI. Apparent mineralocorticoid excess. Trends Endocrinol Metab. 2001; 12:104–111. PMID: 11306334.
Article13. Monder C, Shackleton CH, Bradlow HL, New MI, Stoner E, Iohan F, Lakshmi V. The syndrome of apparent mineralocorticoid excess: its association with 11 beta-dehydrogenase and 5 beta-reductase deficiency and some consequences for corticosteroid metabolism. J Clin Endocrinol Metab. 1986; 63:550–557. PMID: 3460996.14. Stewart PM, Corrie JE, Shackleton CH, Edwards CR. Syndrome of apparent mineralocorticoid excess. A defect in the cortisol-cortisone shuttle. J Clin Invest. 1988; 82:340–349. PMID: 3164727.
Article15. Palermo M, Cossu M, Shackleton CH. Cure of apparent mineralocorticoid excess by kidney transplantation. N Engl J Med. 1998; 339:1787–1788. PMID: 9867558.
Article16. Stewart PM, Murry BA, Mason JI. Human kidney 11 beta-hydroxysteroid dehydrogenase is a high affinity nicotinamide adenine dinucleotide-dependent enzyme and differs from the cloned type I isoform. J Clin Endocrinol Metab. 1994; 79:480–484. PMID: 8045966.
Article17. Stewart PM, Walker BR, Holder G, O'Halloran D, Shackleton CH. 11 beta-Hydroxysteroid dehydrogenase activity in Cushing's syndrome: explaining the mineralocorticoid excess state of the ectopic adrenocorticotropin syndrome. J Clin Endocrinol Metab. 1995; 80:3617–3620. PMID: 8530609.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Hypokalemic Periodic Paralysis Developed in a Patient with Neurogenic Diabetes Insipidus
- Hypokalemic Paralysis: A Report of Two Different Cases
- A Case of Primary Sjiigren's Syndrome with Hypokalemic Paralysis and Renal Tubular Acidosis
- Hypokalemic Periodic Paralysis
- Persistent Hypokalemic Paralysis in a Patient with Graves’ Disease and Gitelman Syndrome
