Yonsei Med J.
2011 Mar;52(2):263-267. 10.3349/ymj.2011.52.2.263.
Changes in Glycogen and Glycosaminoglycan Levels in Hepatocytes of Iduronate-2-Sulfatase Knockout Mice before and after Recombinant Iduronate-2-Sulfatase Supplementation
- Affiliations
-
- 1Department of Pediatrics, Kangnam Sacred Heart Hospital, Hallym University School of Medicine, Seoul, Korea.
- 2Department of Pediatrics, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea. jindk@skku.edu
- KMID: 1779661
- DOI: http://doi.org/10.3349/ymj.2011.52.2.263
Abstract
- PURPOSE
Mucopolysaccharidosis II (MPS II) is a lysosomal storage disorder caused by a deficiency of iduronate-2 sulfatase (IdS), which is involved in the degradation of glycosaminoglycan (GAG). In this study, the frequency of fasting hypoglycemia in patients with MPS II was investigated and changes in accumulation of glycogen and GAG in the hepatocytes of IdS-knockout (KO) mice were evaluated before and after recombinant IdS enzyme replacement therapy (ERT).
MATERIALS AND METHODS
Plasma glucose levels were evaluated after an 8-hour fast in 50 patients with MPS II. The IdS-KO mice were divided into three groups (group 2; saline, group 3; 0.15 mg/kg of IdS, and group 4; 0.5 mg/kg of IdS); wild-type mice were included as controls (group 1). ERT was initiated intravenously at four weeks of age, and continued every week until 20 weeks of age.
RESULTS
The mean glucose level after an 8-hour fast was 94.1 +/- 23.7 mg/dL in the patients with MPS II. Two (4%) out of 50 patients had fasting hypoglycemia. For the mice, GAG in the lysosomes nearly disappeared and glycogen particles in the cytoplasm were restored to the normal range in group 4.
CONCLUSION
Glucose metabolism in patients with MPS II appeared to function well despite hepatocytic GAG accumulation and hypothetical glycogen depletion. A higher dose of IdS infusion in MPS II mice led to disappearance of lysosomal GAG and restoration of glycogen to the cytoplasm of hepatocytes.
Keyword
MeSH Terms
-
Animals
Blood Glucose/analysis
Enzyme Replacement Therapy/methods
Glycogen/*analysis
Glycosaminoglycans/*analysis
Hepatocytes/chemistry/*enzymology
Humans
Hypoglycemia/enzymology/physiopathology
Iduronate Sulfatase/genetics/*physiology
Liver/ultrastructure
Mice
Mice, Knockout
Microscopy, Electron
Mucopolysaccharidosis II/blood/enzymology/physiopathology
Figure
-
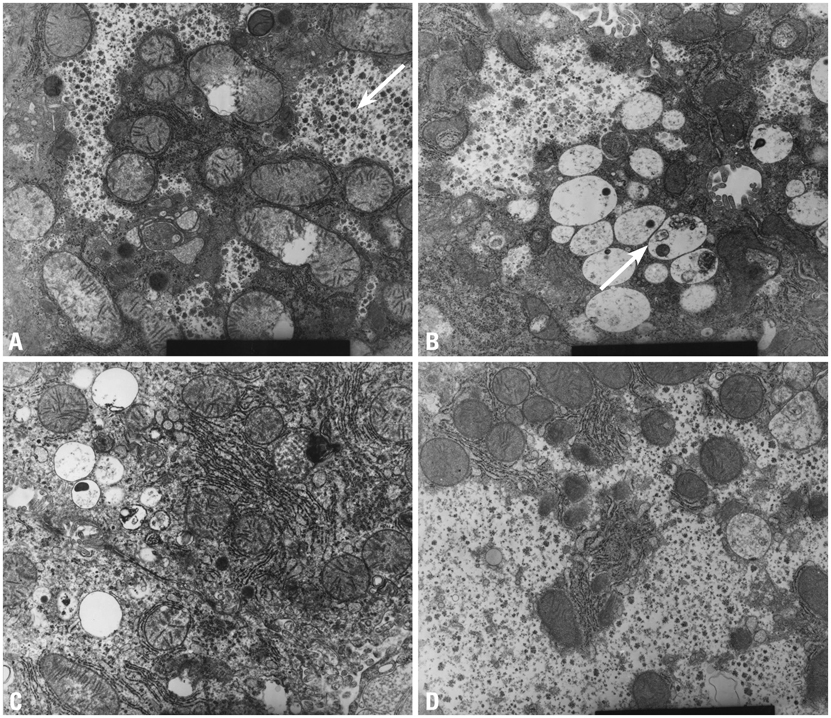
Fig. 1 High power electron microscopic findings (9.0 × 9,000). (A) Wild type: the arrow shows large amounts of glycogen particles in the cytoplasm. (B) MPS II model: the arrow shows accumulation of GAG in lysosomes and scanty glycogen content in the cytoplasm. (C) Low dose ERT group: GAG in the lysosomes decreases but restoration of glycogen in the cytoplasm is not remarkable. (D) High dose ERT group: GAG in the lysosomes nearly disappears and glycogen particles in the cytoplasm are restored. MPS II, mucopolysaccharidosis II; GAG, glycosaminoglycan; ERT, enzyme replacement therapy.
Reference
-
1. Neufeld EF, Muenzer J. Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, editors. The mucopolysaccharidoses. The Metabolic and Molecular Bases of Inherited Disease. 2001. 8th ed. New York: McGraw-Hill;3421–3452.2. Muenzer J, Lamsa JC, Garcia A, Dacosta J, Garcia J, Treco DA. Enzyme replacement therapy in mucopolysaccharidosis type II (Hunter syndrome): a preliminary report. Acta Paediatr Suppl. 2002. 91:98–99.
Article3. Muenzer J, Wraith JE, Beck M, Giugliani R, Harmatz P, Eng CM, et al. A phase II/III clinical study of enzyme replacement therapy with idursulfase in mucopolysaccharidosis II (Hunter syndrome). Genet Med. 2006. 8:465–473.
Article4. Muenzer J, Gucsavas-Calikoglu M, McCandless SE, Schuetz TJ, Kimura A. A phase I/II clinical trial of enzyme replacement therapy in mucopolysaccharidosis II (Hunter syndrome). Mol Genet Metab. 2007. 90:329–337.
Article5. Garcia AR, DaCosta JM, Pan J, Muenzer J, Lamsa JC. Preclinical dose ranging studies for enzyme replacement therapy with idursulfase in a knock-out mouse model of MPS II. Mol Genet Metab. 2007. 91:183–190.
Article6. Huidekoper HH, Duran M, Turkenburg M, Ackermans MT, Sauerwein HP, Wijburg FA. Fasting adaptation in idiopathic ketotic hypoglycemia: a mismatch between glucose production and demand. Eur J Pediatr. 2008. 167:859–865.
Article7. Jung SC, Park ES, Choi EN, Kim CH, Kim SJ, Jin DK. Characterization of a novel mucopolysaccharidosis type II mouse model and recombinant AAV2/8 vector-mediated gene therapy. Mol Cells. 2010. 30:13–18.
Article8. Resnick JM, Whitley CB, Leonard AS, Krivit W, Snover DC. Light and electron microscopic features of the liver in mucopolysaccharidosis. Hum Pathol. 1994. 25:276–286.
Article9. Parfrey NA, Hutchins GM. Hepatic fibrosis in the mucopolysaccharidoses. Am J Med. 1986. 81:825–829.
Article10. Yoshimoto T, Nakamuta M, Kotoh K, Kohjima M, Morizono S, Miyagi Y, et al. An adult case with Hunter's syndrome presenting prominent hepatic failure: light and electron microscopic features of the liver. Intern Med. 2006. 45:1133–1135.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- A case of simultaneously identified glycogen storage disease and mucopolysaccharidosis
- A study of the relationship between clinical phenotypes and plasma iduronate-2-sulfatase enzyme activities in Hunter syndrome patients
- Impact of Enzyme Replacement Therapy on Linear Growth in Korean Patients with Mucopolysaccharidosis Type II (Hunter Syndrome)
- Birth of a healthy baby after preimplantation genetic diagnosis in a carrier of mucopolysaccharidosis type II: The first case in Korea
- Diagnosis of x-linked ichthyosis and detection of its carriers with southern blot hybidization
