Short-Term Efficacy of Enzyme Replacement Therapy in Korean Patients with Fabry Disease
- Affiliations
-
- 1Department of Pediatrics, Chungnam National University Hospital, College of Medicine, Chungnam National University, Daejeon, Korea.
- 2Department of Pathology, Research Institute for Medical Sciences, Chungnam National University Hospital, College of Medicine, Chungnam National University, Daejeon, Korea.
- 3Department of Pathology, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea.
- 4Medical Genetics Clinic and Laboratory, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea.
- 5Department of Pediatrics, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea. hwyoo@amc.seoul.kr
- 6Biomedical and Pharmaceutical Analysis Laboratory, Department of Analytical Chemistry, School of Pharmacy, Duksung Women's University, Seoul, Korea.
- KMID: 1713456
- DOI: http://doi.org/10.3346/jkms.2008.23.2.243
Abstract
- Fabrazyme has been widely used for treatment of Fabry disease since its approval by the U.S. Food and Drug Administration in 2003. This study was undertaken to assess the short-term efficacy and safety of enzyme replacement therapy (ERT) for Fabry disease in Korea. Eight male patients and three female symptomatic carriers aged 13 to 48 yr were included. Fabrazyme was administered by intravenous infusion at a dose of 1 mg/kg every 2 weeks. Plasma and urine globotriaosylceramide (GL-3) levels, serum creatinine, creatinine clearance, and 24-hr urine protein levels were measured every 3 months. Kidney biopsies, ophthalmologic exams, and pure tone audiometry were performed before and 1 yr after ERT. Kidney function, including serum creatinine, creatinine clearance, and the 24-hr urine protein level, remained stable during ERT. Plasma and urine GL-3 levels were reduced within 3 to 6 months of ERT initiation. Microvascular endothelial deposits of GL-3 were decreased from renal biopsy specimens after 1 yr of treatment. The severity of sensorineural hearing loss and tinnitus did not improve after ERT. ERT is safe and effective in stabilizing renal function and clearing microvascular endothelial GL-3 from kidney biopsy specimen in Korean patients with Fabry disease.
Keyword
MeSH Terms
Figure
-
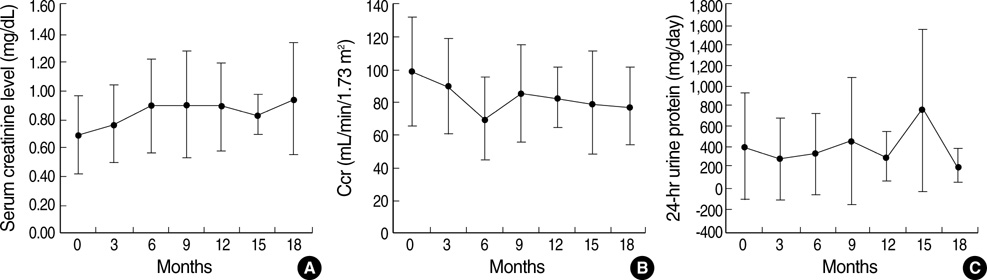
Fig. 1 Renal function of patients during enzyme replacement therapy. Serum creatinine (A), creatinine clearance (B), and 24-hr urine protein levels (C) remained stable without aggravation or improvement (p>0.05). Data are presented as mean±SD.
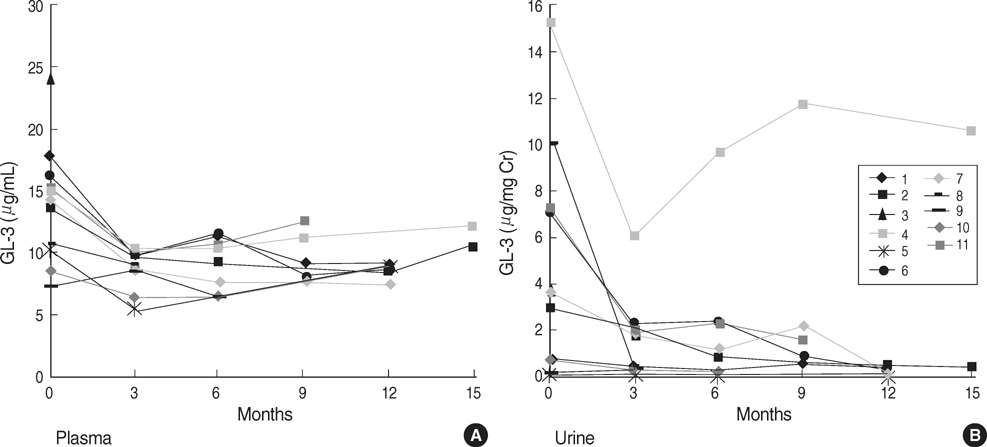
Fig. 2 Sequential changes in the GL-3 concentration in plasma (A) and urine (B) during enzyme replacement therapy.
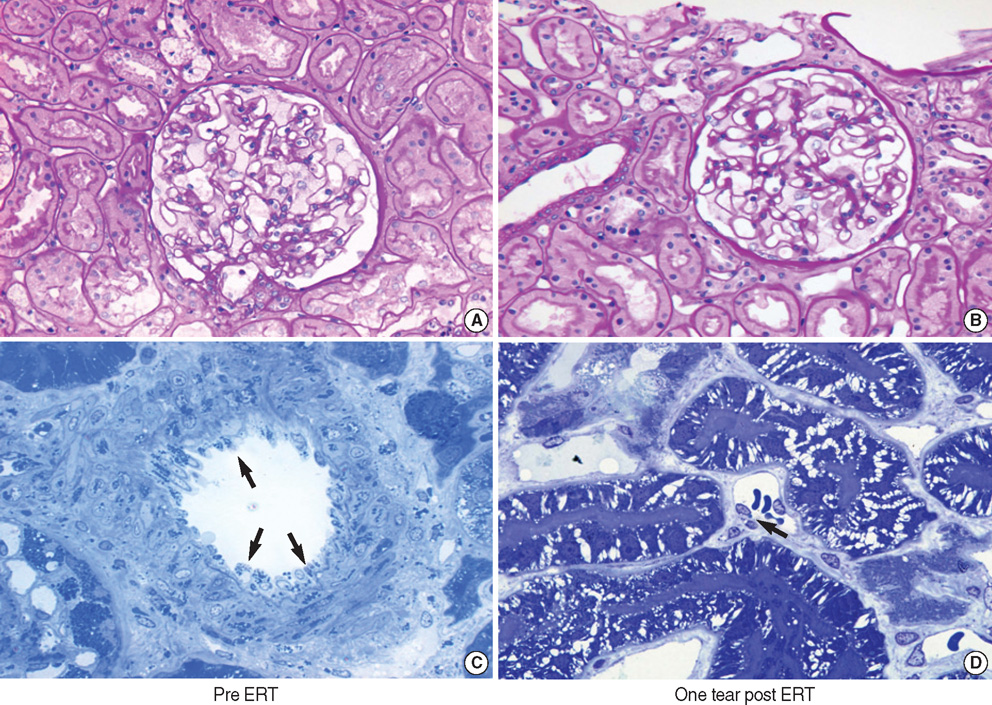
Fig. 3 Pathologic features before (A, C) and after (B, D) ERT. No significant changes were identified the glomerulus and tubulointerstitium on histologic examination. However, the vascular endothelial GL-3 depositions (arrows) were markedly decreased after the ERT (from patient 1; A and B, PAS staining at ×400 magnification; C and D, toluidine blue staining of semithin sections at ×1,000 magnification).
Cited by 1 articles
-
Fabry Cardiomyopathy
Jae Yong Yoon, Joon Hyuk Song, Sang Soo Cheon, Hyun Jun Cho, Myung Hwan Bae, Jang Hoon Lee, Dong Heon Yang, Hun Sik Park, Yongkeun Cho, Shung Chull Chae
J Cardiovasc Ultrasound. 2013;21(1):26-29. doi: 10.4250/jcu.2013.21.1.26.
Reference
-
1. Kint JA. Fabry's disease: alpha-galactosidase deficiency. Science. 1970. 167:1268–1269.
Article2. Desnick RJ, Ioannou YA, Eng CM. Scriver CR, Beaudet AL, Sly WS, editors. α-galactosidase A deficiency: Fabry disease. The Metabolic and Molecular Bases of Inherited Disease. 2001. 8th edn. New York: McGraw-Hill;3733–3774.3. Sheth KJ, Roth DA, Adams MB. Early renal failure in Fabry's disease. Am J Kidney Dis. 1983. 2:651–654.
Article4. Peters FP, Sommer A, Vermeulen A, Cheriex EC, Kho TL. Fabry's disease: a multidisciplinary disorder. Postgrad Med J. 1997. 73:710–712.
Article5. Branton MH, Schiffmann R, Sabnis SG, Murray GJ, Quirk JM, Altarescu G, Goldfarb L, Brady RO, Balow JE, Austin Iii HA, Kopp JB. Natural history of Fabry renal disease: influence of alpha-galactosidase A activity and genetic mutations on clinical course. Medicine (Baltimore). 2002. 81:122–138.6. Gupta S, Ries M, Kotsopoulos S, Schiffmann R. The relationship of vascular glycolipid storage to clinical manifestations of Fabry disease: a cross-sectional study of a large cohort of clinically affected heterozygous women. Medicine (Baltimore). 2005. 84:261–268.7. Nakao S, Takenaka T, Maeda M, Kodama C, Tanaka A, Tahara M, Yoshida A, Kuriyama M, Hayashibe H, Sakuraba H, Tanaka H. An atypical variant of Fabry's disease in men with left ventricular hypertrophy. N Engl J Med. 1995. 333:288–293.
Article8. Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA. 1999. 281:249–254.
Article9. Thadhani R, Wolf M, West ML, Tonelli M, Ruthazer R, Pastores GM, Obrador GT. Patients with Fabry disease on dialysis in the United States. Kidney Int. 2002. 61:249–255.
Article10. Eng CM, Banikazemi M, Gordon RE, Goldman M, Phelps R, Kim L, Gass A, Winston J, Dikman S, Fallon JT, Brodie S, Stacy CB, Mehta D, Parsons R, Norton K, O'Callaghan M, Desnick RJ. A phase 1/2 clinical trial of enzyme replacement in Fabry disease: pharmacokinetic, substrate clearance, and safety studies. Am J Hum Genet. 2001. 68:711–722.
Article11. Eng CM, Guffon N, Wilcox WR, Germain DP, Lee P, Waldek S, Caplan L, Linthorst GE, Desnick RJ; International Collaborative Fabry Disease Study Group. Safety and efficacy of recombinant human α-galactosidase A replacement therapy in Fabry's disease. N Engl J Med. 2001. 345:9–16.
Article12. Wilcox WR, Banikazemi M, Guffon N, Waldek S, Lee P, Linthorst GE, Desnick RJ, Germain DP; International Fabry Disease Study Group. Long-term safety and efficacy of enzyme replacement therapy for Fabry disease. Am J Hum Genet. 2004. 75:65–74.13. Schiffmann R, Murray GJ, Treco D, Daniel P, Sellos-Moura M, Myers M, Quirk JM, Zirzow GC, Borowski M, Loveday K, Anderson T, Gillespie F, Oliver KL, Jeffries NO, Doo E, Liang TJ, Kreps C, Gunter K, Frei K, Crutchfield K, Selden RF, Brady RO. Infusion of alpha-galactosidase A reduces tissue globotriaosylceramide storage in patients with Fabry disease. Proc Natl Acad Sci USA. 2000. 97:365–370.14. Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959. 37:911–917.
Article15. Kayganich KA, Murphy RC. Fast atom bombardment tandem mass spectrometric identification of diacyl, alkylacyl, and alk-1-enylacyl molecular species of glycerophosphoethanolamine in human polymorphonuclear leukocytes. Anal Chem. 1992. 64:2965–2971.
Article16. Blanch LC, Meaney C, Morris CP. A sensitive mutation screening strategy for Fabry disease: detection of nine mutations in the alpha-galactosidase A gene. Hum Mutat. 1996. 8:38–43.17. MacDermot KD, Holmes A, Miners AH. Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 98 hemizygous males. J Med Genet. 2001. 38:750–760.
Article18. Whybra C, Kampmann C, Willers I, Davies J, Winchester B, Kriegsmann J, Bruhl K, Gal A, Bunge S, Beck M. Anderson-Fabry disease: clinical manifestations of disease in female heterozygotes. J Inherit Metab Dis. 2001. 24:715–724.
Article19. Yoshitama T, Nakao S, Takenaka T, Teraguchi H, Sasaki T, Kodama C, Tanaka A, Kisanuki A, Tei C. Molecular genetic, biochemical, and clinical studies in three families with cardiac Fabry's disease. Am J Cardiol. 2001. 87:71–75.
Article20. Desnick RJ, Brady RO. Fabry disease in childhood. J Pediatr. 2004. 144:5 Suppl. S20–S26.
Article21. Kornreich R, Desnick RJ, Bishop DF. Nucleotide sequence of the human alpha-galactosidase A gene. Nucleic Acids Res. 1989. 17:3301–3302.22. Lee JK, Kim GH, Kim JS, Kim KK, Lee MC, Yoo HW. Identification of four novel mutations in five unrelated Korean families with Fabry disease. Clin Genet. 2000. 58:228–233.
Article23. Schiffmann R, Kopp JB, Austin HA 3rd, Sabnis S, Moore DF, Weibel T, Balow JE, Brady RO. Enzyme replacement therapy in Fabry disease: a randomized controlled trial. JAMA. 2001. 285:2743–2749.24. Schiffmann R, Floeter MK, Dambrosia JM, Gupta S, Moore DF, Sharabi Y, Khurana RK, Brady RO. Enzyme replacement therapy improves peripheral nerve and sweat function in Fabry disease. Muscle Nerve. 2003. 28:703–710.
Article25. Whitfield PD, Calvin J, Hogg S, O'Driscoll E, Halsall D, Burling K, Maguire G, Wright N, Cox TM, Meikle PJ, Deegan PB. Monitoring enzyme replacement therapy in Fabry disease-role of urine globotriaosylceramide. J Inherit Metab Dis. 2005. 28:21–33.
Article26. Desnick RJ, Brady R, Barranger J, Collins AJ, Germain DP, Goldman M, Grabowski G, Packman S, Wilcox WR. Fabry disease, an under-recognized multisystemic disorder: expert recommendations for diagnosis, management, and enzyme replacement therapy. Ann Intern Med. 2003. 138:338–346.
Article27. Ries M, Gupta S, Moore DF, Sachdev V, Quirk JM, Murray GJ, Rosing DR, Robinson C, Schaefer E, Gal A, Dambrosia JM, Garman SC, Brady RO, Schiffmann R. Pediatric Fabry disease. Pediatrics. 2005. 115:e344–e355.
Article28. Ries M, Ramaswami U, Parini R, Lindblad B, Whybra C, Willers I, Gal A, Beck M. The early clinical phenotype of Fabry disease: a study on 35 European children and adolescents. Eur J Pediatr. 2003. 162:767–772.
Article29. Ries M, Clarke JT, Whybra C, Timmons M, Robinson C, Schlaggar BL, Pastores G, Lien YH, Kampmann C, Brady RO, Beck M, Schiffmann R. Enzyme-replacement therapy with agalsidase alfa in children with Fabry disease. Pediatrics. 2006. 118:924–932.
Article30. Pisani A, Spinelli L, Sabbatini M, Andreucci MV, Procaccini D, Abbaterusso C, Pasquali S, Savoldi S, Comotti C, Cianciaruso B. Enzyme replacement therapy in Fabry disease patients undergoing dialysis: effects on quality of life and organ involvement. Am J Kidney Dis. 2005. 46:120–127.
Article31. Hajioff D, Enever Y, Quiney R, Zuckerman J, Mackermot K, Mehta A. Hearing loss in Fabry disease: the effect of agalsidase alfa replacement therapy. J Inherit Metab Dis. 2003. 26:787–794.
Article32. Hajioff D, Hegemannn S, Conti G, Beck M, Sunder-Plassmann G, Widmer U, Mehta A, Keilmann A. Agalsidase alpha and hearing in Fabry disease: data from the Fabry Outcome Survey. Eur J Clin Invest. 2006. 36:663–667.
Article33. Wilcox WR. Lysosomal storage disorders: the need for better pediatric recognition and comprehensive care. J Pediatr. 2004. 144:5 Suppl. S3–S14.
Article34. Mignani R, Panichi V, Giudicissi A, Taccola D, Boscaro F, Feletti C, Moneti G, Cagnoli L. Enzyme replacement therapy with agalsidase beta in kidney transplant patients with Fabry disease: a pilot study. Kidney Int. 2004. 65:1381–1385.
Article35. Brooks DA, Kakavanos R, Hopwood JJ. Significance of immune response to enzyme-replacement therapy for patients with a lysosomal storage disorder. Trends Mol Med. 2003. 9:450–453.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Two cases of Fabry disease identified in brothers
- Two Cases of Fabry Disease in Women with Proteinuria Diagnosed by Molecular Analysis of the alpha-Galactosidase A Gene and Kidney Biopsy
- Fabry nephropathy before and after enzyme replacement therapy: important role of renal biopsy in patients with Fabry disease
- Early Diagnosis of Fabry Disease in a Patient with Toe Tip Pain
- A Case of Fabry Disease, Pathologically Revealed as Focal Segmental Glomerulosclerosis
