Ann Lab Med.
2013 Jul;33(4):293-296. 10.3343/alm.2013.33.4.293.
A Novel UMOD Mutation (c.187T>C) in a Korean Family with Juvenile Hyperuricemic Nephropathy
- Affiliations
-
- 1Department of Laboratory Medicine and Genetics, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea. changski@skku.edu
- 2Department of Medicine, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea. wooseong.huh@samsung.com
- 3Department of Pathology, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea.
- KMID: 1707239
- DOI: http://doi.org/10.3343/alm.2013.33.4.293
Abstract
- Familial juvenile hyperuricemic nephropathy (FJHN; OMIM 162000) is an autosomal dominant disorder characterized by hyperuricemia and gouty arthritis due to reduced kidney excretion of uric acid and progressive renal failure. Gradual progressive interstitial renal disease, with basement membrane thickening and glomerulosclerosis resulting from fibrosis, starts in early life. In most cases of FJHN, uromodulin gene (UMOD) is responsible for the disease; however, there has been only one report of a genetically confirmed FJHN family in Korea. Here we report another Korean family with FJHN, in which three male members. a father and 2 sons.developed gout and progressive renal insufficiency. The clinical, laboratory, and radiological findings were consistent with FJHN, and renal biopsy showed chronic parenchymal damage, which can be found in FJHN but is not specific to this disease. In order to confirm the diagnosis, sequence analysis of the UMOD was performed, and a novel heterozygous missense variant (c.187T>C; p.Cys63Arg) in exon 3 was identified. We assume that this variant is likely to be the causative mutation in this family, as the variant segregated with the disease. In addition, approximately two-thirds of the known mutations lead to a cysteine amino acid change in uromodulin, and all such variants have been shown to cause UMOD-associated kidney disease. In summary, we report a Korean FJHN family with three affected members by genetic analysis of the UMOD, and provide the first report of a novel heterozygous missense mutation.
MeSH Terms
Figure
-
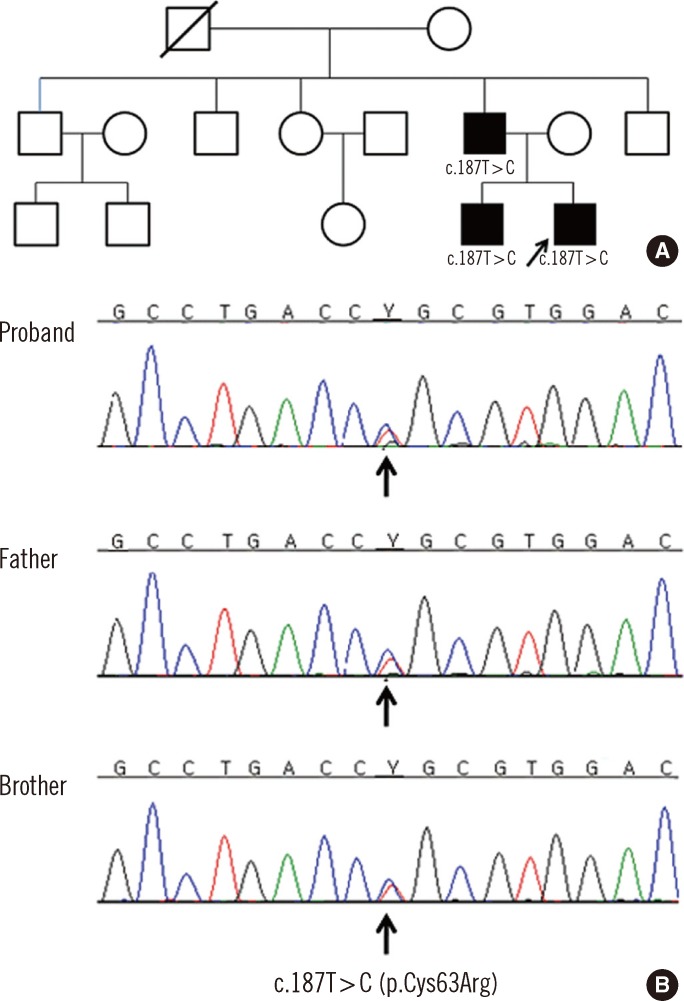
Fig. 1 Pedigree and sequence analysis data from a Korean family with juvenile hyperuricemic nephropathy (FJHN). (A) Pedigree of the family. The proband is indicated by an arrow. Open symbols indicate no signs or symptoms of FJHN. Filled symbols represent affected individuals and deceased individual is crossed. Genotypes are shown for the individuals with FJHN. (B) Sequence analysis of the UMOD revealed that the proband, his father, and his elder brother were heterozygous for a novel mutation in the UMOD. The arrow indicates the overlapping peaks at nucleotide position 187, due to a heterozygous T>C substitution (c.187T>C; p.Cys63Arg).
Reference
-
1. Duncan H, Dixon AS. Gout, familial hypericaemia, and renal disease. Q J Med. 1960; 29:127–135. PMID: 13818629.2. Vylet'al P, Kublová M, Kalbácova M, Hodanová K, Baresová V, Stibůrková B, et al. Alterations of uromodulin biology: a common denominator of the genetically heterogeneous FJHN/MCKD syndrome. Kidney Int. 2006; 70:1155–1169. PMID: 16883323.3. Hart TC, Gorry MC, Hart PS, Woodard AS, Shihabi Z, Sandhu J, et al. Mutations of the UMOD are responsible for medullary cystic kidney disease 2 and familial juvenile hyperuricaemic nephropathy. J Med Genet. 2002; 39:882–892. PMID: 12471200.4. Turner JJ, Stacey JM, Harding B, Kotanko P, Lhotta K, Puig JG, et al. UROMODULIN mutations cause familial juvenile hyperuricemic nephropathy. J Clin Endocrinol Metab. 2003; 88:1398–1401. PMID: 12629136.
Article5. Pook MA, Jeremiah S, Scheinman SJ, Povey S, Thakker RV. Localization of the Tamm-Horsfall glycoprotein (uromodulin) gene to chromosome 16p12.3-16p13.11. Ann Hum Genet. 1993; 57:285–290. PMID: 8179291.6. Bingham C, Ellard S, van't Hoff WG, Simmonds HA, Marinaki AM, Badman MK, et al. Atypical familial juvenile hyperuricemic nephropathy associated with a hepatocyte nuclear factor-1beta gene mutation. Kidney Int. 2003; 63:1645–1651. PMID: 12675839.7. Edghill EL, Bingham C, Ellard S, Hattersley AT. Mutations in hepatocyte nuclear factor-1beta and their related phenotypes. J Med Genet. 2006; 43:84–90. PMID: 15930087.8. Hodanová K, Majewski J, Kublová M, Vyletal P, Kalbácova M, Stibůrková B, et al. Mapping of a new candidate locus for uromodulin-associated kidney disease (UAKD) to chromosome 1q41. Kidney Int. 2005; 68:1472–1482. PMID: 16164624.9. Rampoldi L, Caridi G, Santon D, Boaretto F, Bernascone I, Lamorte G, et al. Allelism of MCKD, FJHN and GCKD caused by impairment of uromodulin export dynamics. Hum Mol Genet. 2003; 12:3369–3384. PMID: 14570709.
Article10. Lens XM, Banet JF, Outeda P, Barrio-Lucia V. A novel pattern of mutation in uromodulin disorders: autosomal dominant medullary cystic kidney disease type 2, familial juvenile hyperuricemic nephropathy, and autosomal dominant glomerulocystic kidney disease. Am J Kidney Dis. 2005; 46:52–57. PMID: 15983957.
Article11. Pennica D, Kohr WJ, Kuang WJ, Glaister D, Aggarwal BB, Chen EY, et al. Identification of human uromodulin as the Tamm-Horsfall urinary glycoprotein. Science. 1987; 236:83–88. PMID: 3453112.
Article12. Kumar S, Muchmore A. Tamm-Horsfall protein--uromodulin (1950-1990). Kidney Int. 1990; 37:1395–1401. PMID: 2194064.
Article13. Rindler MJ, Naik SS, Li N, Hoops TC, Peraldi MN. Uromodulin (Tamm-Horsfall glycoprotein/uromucoid) is a phosphatidylinositol-linked membrane protein. J Biol Chem. 1990; 265:20784–20789. PMID: 2249987.
Article14. Serafini-Cessi F, Malagolini N, Cavallone D. Tamm-Horsfall glycoprotein: biology and clinical relevance. Am J Kidney Dis. 2003; 42:658–676. PMID: 14520616.
Article15. Wei X, Xu R, Yang Z, Li Z, Liao Y, Johnson RJ, et al. Novel uromodulin mutation in familial juvenile hyperuricemic nephropathy. Am J Nephrol. 2012; 36:114–120. PMID: 22776760.
Article16. Williams SE, Reed AA, Galvanovskis J, Antignac C, Goodship T, Karet FE, et al. Uromodulin mutations causing familial juvenile hyperuricaemic nephropathy lead to protein maturation defects and retention in the endoplasmic reticulum. Hum Mol Genet. 2009; 18:2963–2974. PMID: 19465746.
Article17. Yang H, Wu C, Zhao S, Guo J. Identification and characterization of D8C, a novel domain present in liver-specific LZP, uromodulin and glycoprotein 2, mutated in familial juvenile hyperuricaemic nephropathy. FEBS Lett. 2004; 578:236–238. PMID: 15589826.
Article18. Lee DH, Kim JK, Oh SE, Noh JW, Lee YK. A case of familial juvenile hyperuricemic nephropathy with novel uromodulin gene mutation, a novel heterozygous missense mutation in Korea. J Korean Med Sci. 2010; 25:1680–1682. PMID: 21060763.
Article19. Dahan K, Devuyst O, Smaers M, Vertommen D, Loute G, Poux JM, et al. A cluster of mutations in the UMOD causes familial juvenile hyperuricemic nephropathy with abnormal expression of uromodulin. J Am Soc Nephrol. 2003; 14:2883–2893. PMID: 14569098.
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- A Case of Familial Juvenile Hyperuricemic Nephropathy with Novel Uromodulin Gene Mutation, a Novel Heterozygous Missense Mutation in Korea
- Familial Juvenile Hyperuricemic Nephropathy and Uromodulin Gene Mutation
- Effects of resveratrol on the inflammatory response and renal injury in hyperuricemic rats
- A Novel Germline Mutation in Exon 10 of the SMAD4 Gene in a Familial Juvenile Polyposis
- A Case of Juvenile Xanthogranuloma Developed in a Neurofibromatosis Child with Family History
