Codon Usage Bias of Human Cytomegalovirus Genes with Different Evolutionary Conservancy
- Affiliations
-
- 1Department of Microbiology, Chungbuk National University, Cheongju, Chungbuk, Korea. chlee@chungbuk.ac.kr
- KMID: 1502000
- DOI: http://doi.org/10.4167/jbv.2013.43.4.317
Abstract
- Human cytomegalovirus (HCMV) is a member of beta-herpesvirus and contains a double-stranded genome with longer than 230 Kbp. HCMV infection of human is mostly asymptomatic, but often causes fatal diseases in immunocompromised people. In this study, codon usages of HCMV genes were analyzed and attempted to correlate with evolutionary conservancy. Core genes are the most conserved genes common among herpesvirus family, beta-herpes genes are common to beta-herpesviruses, and CMV genes are the least conserved found only in CMVs. Core genes had higher codon adaptation index (CAI) and GC content of silent 3rd codon position (GC3s) values and lower effective number of codons (Nc) and Nc/GC3s values than CMV genes. The average length of core genes was statistically longer than CMV genes, and core genes were found to be less varied than CMV genes. beta-herpes genes could be placed between core and CMV genes. Higher CAI and GC3s values along with lower Nc and Nc/GC3s values are suggestive of higher codon usage bias and more adaptation to host cells. Thus it is concluded that core genes of HCMV are more biased in codon usage and adapted to host cells compared to CMV genes.
Keyword
Figure
-

Figure 1. Codon usage bias index (CUI) values of HCMV genes. These graphs represent the distribution of CUI values of 3 groups. Each graph shows (A) CAI, (B) GC3s, (C) Nc, and (D) Nc/GC3s values. CORE genes are indicated by black circles, β-HERPES genes by white circles, and CMV genes by black triangles. Black bars indicate the standard error of the mean. Statistical significance of the difference in mean values was examined by student's t-test.

Figure 2. Nc versus GC3s plots for HCMV gene groups. (A) Scatter plot for all HCMV ORFs. (B) plot for CORE genes. (C) plot for β-HERPES genes. (D) plot for CMV genes. The continuous curve represents the expected Nc values by Wright's formula.
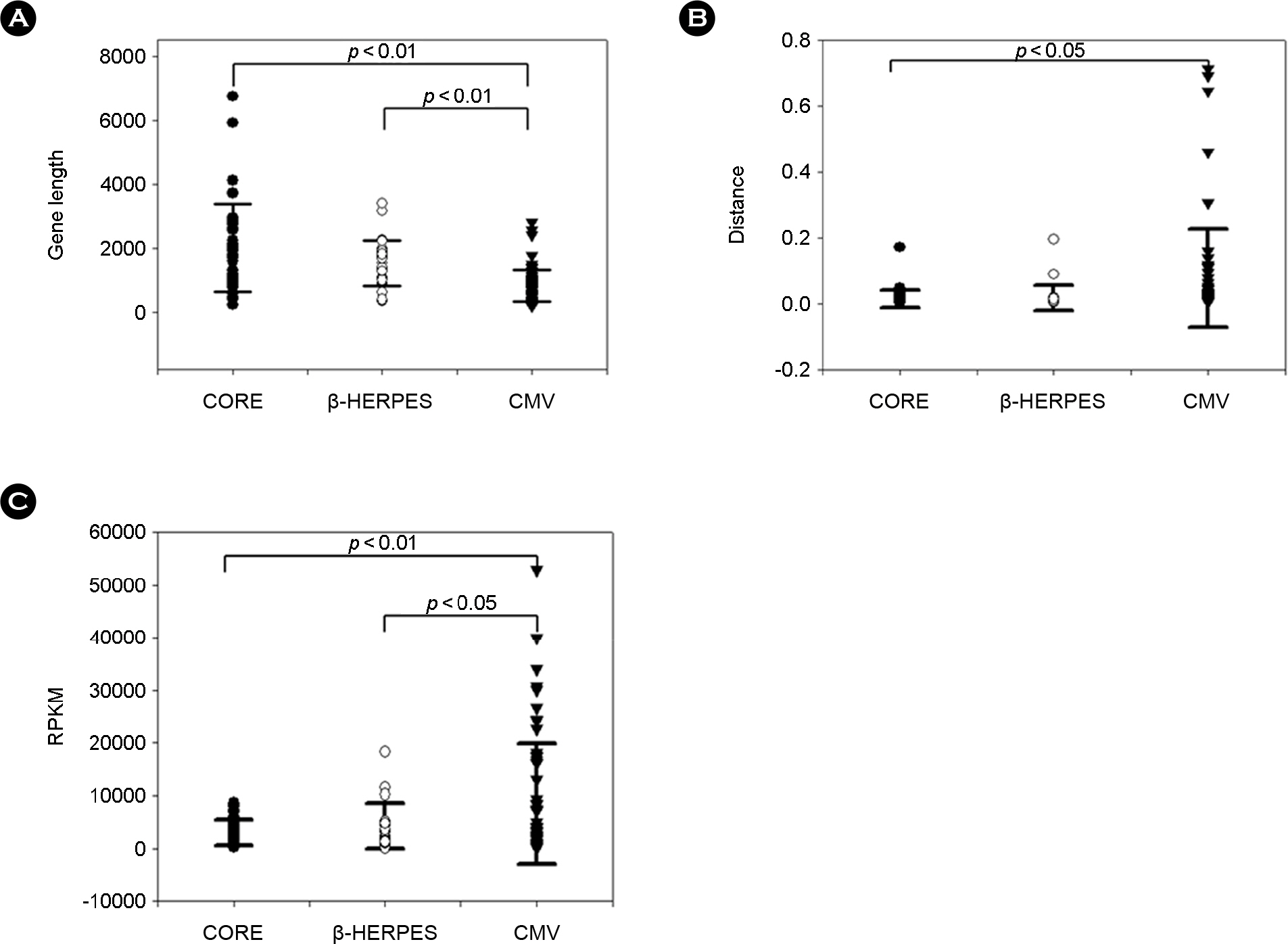
Figure 3. Genetic characteristics of HCMV genes. (A) Distribution of length of HCMV genes. (B) Genetic distance values of HCMV genes. (C) Relative amount of mRNA of HCMV genes. CORE genes are indicated by black circles, β-HERPES genes by white circles, and CMV genes by black triangles. Black bars indicate the standard error of the mean. Statistical significance of the difference in mean values was examined by student's t-test.
Reference
-
1). Landolfo S, Gariglio M, Gribaudo G, Lembo D. The human cytomegalovirus. Pharmacol Ther. 2003; 98:269–97.
Article2). Mocarski ES, Shenk T, Pass RF. Cytomegalovirus. Fields Virology;. 2007. 2702–72.3). Khanna R, Diamond DJ. Human cytomegalovirus vaccine: time to look for alternative options. Trends Mol Med. 2006; 12:26–33.
Article4). Dolan A, Cunningham C, Hector RD, Hassan-Walker AF, Lee L, Addison C, et al. Genetic content of wild-type human cytomegalovirus. J Gen Virol. 2004; 85:1301–12.
Article5). Dunn W, Chou C, Li H, Hai R, Patterson D, Stolc V, et al. Functional profiling of a human cytomegalovirus genome. Proc Natl Acad Sci U S A. 2003; 100:14223–8.
Article6). Rigoutsos I, Novotny J, Huynh T, Chin-Bow ST, Parida L, Platt D, et al. In silico pattern-based analysis of the human cytomegalovirus genome. J Virol. 2003; 77:4326–44.
Article7). Vetsigian K, Goldenfeld N. Genome rhetoric and the emergence of compositional bias. Proc Natl Acad Sci U S A. 2009; 106:215–20.
Article8). Bouquet J, Cherel P, Pavio N. Genetic characterization and codon usage bias of full-length Hepatitis E virus sequences shed new lights on genotypic distribution, host restriction and genome evolution. Infect Genet Evol. 2012; 12:1842–53.
Article9). Shackelton LA, Parrish CR, Holmes EC. Evolutionary basis of codon usage and nucleotide composition bias in vertebrate DNA viruses. J Mol Evol. 2006; 62:551–63.
Article10). Jenkins GM, Holmes EC. The extent of codon usage bias in human RNA viruses and its evolutionary origin. Virus Res. 2003; 92:1–7.
Article11). Karlin S, Blaisdell BE, Schachtel GA. Contrasts in codon usage of latent versus productive genes of Epstein-Barr virus: data and hypotheses. J Virol. 1990; 64:4264–73.
Article12). Fu M. Codon usage bias in herpesvirus. Arch Virol. 2010; 155:391–6.
Article13). Wright F. The ‘effective number of codons’ used in a gene. Gene. 1990; 87:23–9.
Article14). Wu G, Nie L, Zhang W. Predicted highly expressed genes in Nocardia farcinica and the implication for its primary metabolism and nocardial virulence. Antonie Van Leeuwenhoek. 2006; 89:135–46.15). Davison AJ, Dolan A, Akter P, Addison C, Dargan DJ, Alcendor DJ, et al. The human cytomegalovirus genome revisited: comparison with the chimpanzee cytomegalovirus genome. J Gen Virol. 2003; 84:17–28.
Article16). Sharp PM, Li WH. The codon adaptation index –a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987; 15:1281–95.17). Banerjee T, Gupta SK, Ghosh TC. Towards a resolution on the inherent methodological weakness of the “effective number of codons used by a gene”. Biochem Biophys Res Commun. 2005; 330:1015–8.
Article18). Roymondal U, Das S, Sahoo S. Predicting gene expression level from relative codon usage bias: An application to Escherichia coli genome. DNA Res. 2009; 16:13–30.19). Waldman YY, Tuller T, Shlomi T, Sharan R, Ruppin E. Translation efficiency in human: tissue specificity, global optimization and differences between developmental stages. Nucleic Acids Res. 2010; 38:2964–74.20). Goetz RM, Fuglsang A. Correlation of codon bias measures with mRNA levels: analysis of transcriptome data from Escherichia coli. Biochem Biophys Res Commun. 2005; 327:4–7.21). Ohama T, Muto A, Osawa S. Role of GC-biased mutation pressure on synonymous codon choice in Micrococcus luteus, a bacterium with a high genomic GC-content. Nucleic Acids Res. 1990; 18:1565–9.22). Lafay B, Atherton JC, Sharp PM. Absence of translationally selected synonymous codon usage bias in Helicobacter pylori. Microbiology. 2000; 146:851–60.23). dos Reis M, Wernisch L, Savva R. Unexpected correlations between gene expression and codon usage bias from microarray data for the whole Escherichia coli K-12 genome. Nucleic Acids Res. 2003; 31:6976–85.24). Belalov IS, Lukashev AN. Causes and implications of codon usage bias in RNA viruses. PLoS ONE. 2013; 8:e56642.
Article25). Bulmer M. Coevolution of codon usage and transfer RNA abundance. Nature. 1987; 325:728–30.
Article26). Sharp PM, Stenico M, Peden JF, Lloyd AT. Codon usage: mutational bias, translational selection, or both? Biochem Soc Trans. 1993; 21:835–41.
Article27). Jia R, Cheng A, Wang M, Xin H, Guo Y, Zhu D, et al. Analysis of synonymous codon usage in the UL24 gene of duck enteritis virus. Virus Genes. 2009; 38:96–103.
Article28). Jiang Y, Deng F, Wang H, Hu Z. An extensive analysis on the global codon usage pattern of baculoviruses. Arch Virol. 2008; 153:2273–82.
Article29). Hassan S, Mahalingam V, Kumar V. Synonymous codon usage analysis of thirty two mycobacteriophage genomes. Adv Bioinformatics. 2009; 316936.
Article30). Prabha R, Singh DP, Gupta SK, Farooqi S, Rai A. Synonymous codon usage in Thermosynechococcus elongatus (cyanobacteria) identifies the factors shaping codon usage variation. Bioinformation. 2012; 8:622–8.31). Zhao S, Zhang Q, Chen Z, Zhao Y, Zhong J. The factors shaping synonymous codon usage in the genome of Burkholderia mallei. J Genet Genomics. 2007; 34:362–72.
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Codon Usage Patterns of Tyrosinase Genes in Clonorchis sinensis
- Comparative Analysis of Predicted Gene Expression among Crenarchaeal Genomes
- The pattern of coding sequences in the chloroplast genome of Atropa belladonna and a comparative analysis with other related genomes in the nightshade family
- Synonymous Codon Usage Controls Various Molecular Aspects
- Codon Usage Bias and Determining Forces in Taenia solium Genome
