Lab Med Online.
2011 Apr;1(2):115-119. 10.3343/lmo.2011.1.2.9.
Identification of a Novel Mutation in the MCCC2 Gene of a Korean Patient with 3-Methylcrotonyl-CoA Carboxylase Deficiency
- Affiliations
-
- 1Department of Laboratory Medicine, Soonchunhyang University Seoul Hospital and Soonchunhyang University College of Medicine, Seoul, Korea.
- 2Department of Pediatrics, Soonchunhyang University Seoul Hospital and Soonchunhyang University College of Medicine, Seoul, Korea.
- 3Department of Laboratory Medicine and Genetics, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea.
- 4Department of Laboratory Medicine and Genetics, Soonchunhyang University Bucheon Hospital and Soonchunhyang University College of Medicine, Bucheon, Korea. lywmd@schmc.ac.kr
- KMID: 1446279
- DOI: http://doi.org/10.3343/lmo.2011.1.2.9
Abstract
- 3-methylcrotonyl-CoA carboxylase deficiency is an autosomal recessive disorder characterized by a defect in leucine catabolism. We report the case of an 80-day-old patient with 3-methylcrotonyl-CoA carboxylase deficiency who had elevated levels of 3-hydroxyisovalerylcarnitine (45.56 micromol/L; reference range, <0.65 micromol/L), which was detected using tandem mass spectrometry during newborn screening, and elevated levels of 3-hydroxyisovaleric acid (375.75 mmol/mol Cr) and 3-methylcrotonylglycine (502.36 mmol/mol Cr ), which were detected in urine organic acid analysis. We performed direct sequence analysis of all the exons of the MCCC1 and MCCC2 genes. No mutations were detected in the direct sequence analysis of MCCC1. However sequencing of the MCCC2 gene revealed a mutation caused by a heterozygous G to C transversion [c.313G>C (p.Gly105Arg)] at nucleotide position 313 and a mutation caused by a heterozygous A to T transversion [c.1252A>T (p.lle418Phe)] at nucleotide position 1252. Identification of these 2 novel MCCC2 gene mutations in our patient suggested that analysis of the MCCC1 and MCCC2 genes might prove useful in the diagnosis of 3-methylcrotonyl-CoA carboxylase deficiency.
Keyword
MeSH Terms
Figure
-

Fig. 1 Urine organic acid analysis of an 80-day-old patient who had 3-methylcrotonyl-CoA carboxylase deficiency and elevated levels of 3-hydroxyisovaleric acid and 3-methylcrotonylglycine.
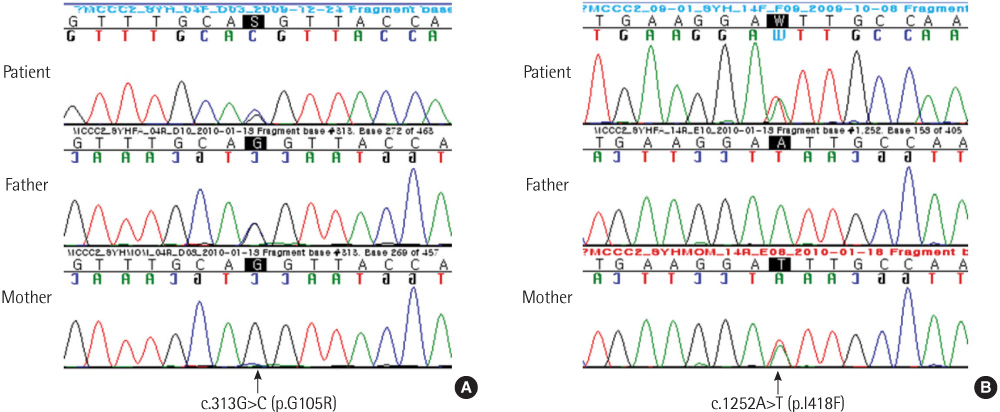
Fig. 2 Mutation analysis of the MCCC2 gene in a Korean patient with 3-methylcrotonyl-CoA carboxylase deficiency. Direct sequencing of the MCCC2 gene shows overlapping peaks (arrow) at nucleotide position 313 because of a heterozygous G to C transversion [c.313G>C (p.Gly105Arg)] (A) and at nucleotide position 1252 because of a heterozygous A to T transversion [c.1252A>T (p.lle418Phe)] (B).
Reference
-
1. Stadler SC, Polanetz R, Maier EM, Heidenreich SC, Niederer B, Mayerhofer PU, et al. Newborn screening for 3-methylcrotonyl-CoA carboxylase deficiency: population heterogeneity of MCCA and MCCB mutations and impact on risk assessment. Hum Mutat. 2006. 27:748–759.
Article2. Naylor EW, Chace DH. Automated tandem mass spectrometry for mass newborn screening for disorders in fatty acid, organic acid and amino acid metabolism. J Child Neurol. 1999. 14:S4–S8.
Article3. Gibson KM, Bennett MJ, Naylor EW, Morton DH. 3-Methylcrotonyl-coenyme A carboxylase deficiency in Amish/Mennonite adults identified by detection of increased acylcarnitines in blood spots of their children. J Pediatr. 1998. 132:519–523.
Article4. Smith WE, Muenzer J, Frazier D, Millington DS, Kishnani PS, Mcdonald M, et al. Evaluation of elevated hydroxyisovalerylcarnitine in the newborn screen by tandem mass spectrometry. Am J Hum Genet. 2000. 67:292.5. Arnold GL, Koeberl DD, Matern D, Barshop B, Braverman N, Burton B, et al. A delphi-based consensus clinical practice protocol for the diagnosis and management of 3-methylcrotonyl CoA carboxylase deficiency. Mol Genet Metab. 2008. 93:363–370.
Article6. Uematsu M, Sakamoto O, Sugawara N, Kumagai N, Morimoto T, Yamaguchi S, et al. Novel mutations in five Japanese patients with 3-methylcrotonyl-CoA carboxylase deficiency. J Hum Genet. 2007. 52:1040–1043.
Article7. Kim JK. A case of asymptomatic 3-methylcrotonylglycinuria detected by tandem mass spectrometry in newborn screening. Korean J Pediatr. 2004. 47:912–916.8. Lehnert W, Niederhoff H, Suormala T, Baumgartner ER. Isolated biotin-resistant 3-methylcrotonyl-CoA carboxylase deficiency: long-term outcome in a case with neonatal onset. Eur J Pediatr. 1996. 155:568–572.
Article9. Murayama K, Kimura M, Yamaguchi S, Shinka T, Kodama K. Isolated 3-methylcrotonyl-CoA carboxylase deficiency in a 15-year-old girl. Brain Dev. 1997. 19:303–305.10. Bartlett K, Bennett MJ, Hill RP, Lashford LS, Pollitt RJ, Worth HG. Isolated biotin-resistant 3-methylcrotonyl CoA carboxylase deficiency presenting with life-threatening hypoglycaemia. J Inherit Metab Dis. 1984. 7:182.
Article11. Gitzelmann R, Steinmann B, Niederwieser A, Fanconi S, Suormala T, Baumgartner ER. Isolated biotin-resistant 3-methylcrotonyl CoA carboxylase deficiency presenting at age 20 months with sopor, hypoglycaemia and ketoacidosis. J Inherit Metab Dis. 1987. 10:290–292.
Article12. Wilcken B, Wiley V, Hammond J, Carpenter K. Screening newborns for inborn errors of metabolism by tandem mass spectrometry. N Engl J Med. 2003. 348:2304–2312.
Article13. Koeberl DD, Millington DS, Smith WE, Weavil SD, Muenzer J, McCandless SE, et al. Evaluation of 3-methylcrotonyl-CoA carboxylase deficiency detected by tandem mass spectrometry newborn screening. J Inherit Metab Dis. 2003. 26:25–35.
Article14. Eichhorst J, Alcorn J, Lepage J, Etter M, Antonishyn NA, Fitterer B, et al. Elevated neonatal 3-OH isovalerylcarnitine due to breast milk sources in maternal 3-MCC deficiency. Mol Genet Metab. 2010. 101:84–86.
Article15. Yap S, Monavari AA, Thornton P, Naughten E. Late-infantile 3-methylcrotonyl-CoA carboxylase deficiency presenting as global developmental delay. J Inherit Metab Dis. 1998. 21:175–176.
Article16. Lamhonwah AM, Barankiewicz TJ, Willard HF, Mahuran DJ, Quan F, Gravel RA. Isolation of cDNA clones coding for the α and β chains of human propionyl-CoA carboxylase: chromosomal assignments and DNA polymorphisms associated with PCCA and PCCB genes. Proc Natl Acad Sci USA. 1986. 83:4864–4868.
Article17. Gallardo ME, Desviat LR, Rodríguez JM, Esparza-Gordillo J, Pérez-Cerdá C, Pérez B, et al. The molecular basis of 3-methylcrotonylglycinuria, a disorder of leucine catabolsim. Am J Hum Genet. 2001. 68:334–346.
Article18. Eminoglu FT, Ozcelik AA, Okur I, Tumer L, Biberoglu G, Demir E, et al. 3-Methylcrotonyl-CoA carboxylase deficiency: phenotypic variability in a family. J Child Neurol. 2009. 24:478–481.
Article19. Stucki M, Suormala T, Fowler B, Valle D, Baumgartner MR. Cryptic exon activation by disruption of exon splice enhancer: novel mechanism causing 3-methylcrotonyl-CoA carboxylase deficiency. J Biol Chem. 2009. 284:28953–28957.20. Nguyen KV, Naviaux RK, Patra S, Barshop BA, Nyhan WL. Novel mutations in the human MCCA and MCCB gene causing methylcrotonylglycinuria. Mol Genet Metab. 2010. In press.
Article21. Desviat LR, Pérez-Cerdá C, Pérez B, Esparza-Gordillo J, Rodriguez-Pombo P, Penalva MA, et al. Functional analysis of MCCA and MCCB mutations causing methylcrotonylglycinuria. Mol Genet Metab. 2003. 80:315–320.
Article22. Baumgartner MR, Almashanu S, Suormala T, Obie C, Cole RN, Packman S, et al. The molecular basis of human 3-methylcrotonyl-CoA carboxylase deficiency. J Clin Invest. 2001. 107:495–504.
Article23. Dantas MF, Suormala T, Randolph A, Coelho D, Fowler B, Valle D, et al. 3-Methylcrotonyl-CoA carboxylase deficiency: mutation analysis in 28 probands, 9 symptomatic and 19 detected by newborn screening. Hum Mutat. 2005. 26:164.
Article24. Ficicioglu C, Payan I. 3-Methylcrotonyl-CoA carboxylase deficiency: metabolic decompensation in a noncompliant child detected through newborn screening. Pediatrics. 2006. 118:2555–2556.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Novel heterozygous MCCC1 mutations identified in a patient with 3-methylcrotonyl-coenzyme A carboxylase deficiency
- A Case of Asymptomatic 3-methylcrotonylglycinuria Detected by Tandem Mass Spectrometry in Newborn Screening
- Clinical Manifestations, Gene Analysis of Patients with 3-Methylcrotonyl-CoA Carboxylase Deficiency
- Isolated 3-Methylcrotonyl CoA Carboxylase Deficiency Detected by Newborn Screening Program Using Tandem Mass Spectrometry
- A Case of Lennox-Gastaut Syndrome due to 3-Methylcrotonyl CoA Carboxylase Deficiency
