Clinical, Biochemical and Genetic Analyses in Two Korean Patients with Medium-chain Acyl-CoA Dehydrogenase Deficiency
- Affiliations
-
- 1Department of Laboratory Medicine & Genetics, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea.
- 2Department of Laboratory Medicine & Genetics, Soonchunhyang University Bucheon Hospital, Soonchunhyang University College of Medicine, Bucheon, Korea. nayadoo@hanmail.net lywmd@hanmail.net
- 3Department of Pediatrics, College of Medicine, Soonchunhyang University, Seoul, Korea.
- KMID: 1096662
- DOI: http://doi.org/10.3343/kjlm.2011.31.1.54
Abstract
- Medium-chain acyl-CoA dehydrogenase deficiency (MCADD) is an autosomal recessive hereditary metabolic disorder of mitochondrial fatty acid beta-oxidation. It is characterized by hypoketotic hypoglycemia, hyperammonemia, seizure, coma, and sudden infant death syndrome-like illness. The most frequently isolated mutation in the acyl-CoA dehydrogenase, medium-chain (ACADM) gene of Caucasian patients with MCADD is c.985A>G, but ethnic variations exist in the frequency of this mutation. Here, we describe 2 Korean pediatric cases of MCADD, which was detected during newborn screening by tandem mass spectrometry and confirmed by molecular analysis. The levels of medium-chain acylcarnitines, including octanoylcarnitine (C8), hexanoylcarnitine (C6), and decanoylcarnitine (C10), were typically elevated. Molecular studies revealed that Patient 1 was a compound heterozygote for c.449_452delCTGA (p.Thr150ArgfsX4) and c.461T>G (p.L154W) mutations, and Patient 2 was a compound heterozygote for c.449_452delCTGA (p.Thr150ArgfsX4) and c.1189T>A (p.Y397N) mutations. We detected asymptomatic patients with MCADD by using a newborn screening test and confirmed it by ACADM mutation analysis. This report presents evidence of the biochemical and molecular features of MCADD in Korean patients and, to the best of our knowledge, this is the first report of the c.461T>G mutation in the ACADM gene.
MeSH Terms
-
Acyl-CoA Dehydrogenase/chemistry/deficiency/genetics
Asian Continental Ancestry Group/*genetics
Base Sequence
Biological Markers/blood
Carnitine/analogs & derivatives/blood
DNA Mutational Analysis
Exons
Female
Gene Deletion
Heterozygote
Humans
Infant, Newborn
Lipid Metabolism, Inborn Errors/diagnosis/genetics
Male
Mutation
Neonatal Screening
Republic of Korea
Tandem Mass Spectrometry
Figure
-
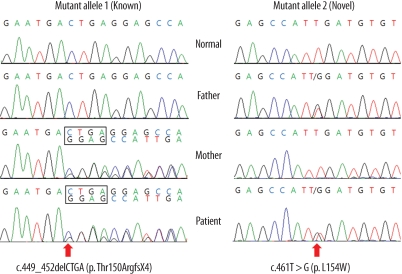
Fig. 1 ACADM mutations detected by sequence analysis. Compound heterozygote consisting of a known mutation and a novel mutation in Patient 1. The known maternally-derived allele was a 4-bp deletion at position 449 to 452 in exon 6, altering codon 153 from a proline to a termination codon (p.Thr150ArgfsX4). The novel mutant allele originated from the patient's father and was a T to G transition at position 461 in exon 6, resulting in a leucine to tryptophan change at codon 154 (p.L154W).
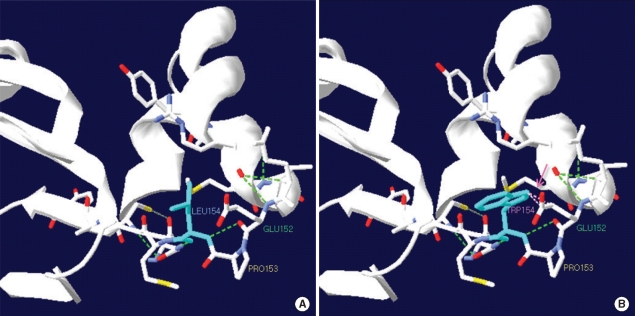
Fig. 2 Diagram of protein structure near the c.461T>G (p.L154W) mutation. (A) Leu154 lies on the loop between the N-terminal α domain and β domain, shown in sky blue. (B) The novel Leu154Trp mutation produces side-chain clash with Glu152 (dotted pink line). Hydrogen bonds are shown by the dotted green lines.
Cited by 1 articles
-
Neonatal Screening Tests for Inherited Metabolic Disorders using Tandem Mass Spectrometry: Experience of a Clinical Laboratory in Korea
Sung Eun Cho, Eun Jung Park, Dong Hee Seo, In Bum Lee, Hyun Ju Lee, Dae-Yeon Cho, Jung Min Oh
Lab Med Online. 2015;5(4):196-203. doi: 10.3343/lmo.2015.5.4.196.
Reference
-
1. Iafolla AK, Thompson RJ Jr, Roe CR. Medium-chain acyl-coenzyme A dehydrogenase deficiency: clinical course in 120 affected children. J Pediatr. 1994; 124:409–415. PMID: 8120710.
Article2. Andresen BS, Dobrowolski SF, O'Reilly L, Muenzer J, McCandless SE, Frazier DM, et al. Medium-chain acyl-CoA dehydrogenase (MCAD) mutations identified by MS/MS-based prospective screening of newborns differ from those observed in patients with clinical symptoms: identification and characterization of a new, prevalent mutation that results in mild MCAD deficiency. Am J Hum Genet. 2001; 68:1408–1418. PMID: 11349232.
Article3. Andresen BS, Bross P, Udvari S, Kirk J, Gray G, Kmoch S, et al. The molecular basis of medium-chain acyl-CoA dehydrogenase (MCAD) deficiency in compound heterozygous patients: is there correlation between genotype and phenotype? Hum Mol Genet. 1997; 6:695–707. PMID: 9158144.
Article4. Sander S, Janzen N, Janetzky B, Scholl S, Steuerwald U, Schäfer J, et al. Neonatal screening for medium chain acyl-CoA deficiency: high incidence in Lower Saxony (northern Germany). Eur J Pediatr. 2001; 160:318–319. PMID: 11388605.
Article5. Maier EM, Liebl B, Röschinger W, Nennstiel-Ratzel U, Fingerhut R, Olgemöller B, et al. Population spectrum of ACADM genotypes correlated to biochemical phenotypes in newborn screening for medium-chain acyl-CoA dehydrogenase deficiency. Hum Mutat. 2005; 25:443–452. PMID: 15832312.6. Grosse SD, Khoury MJ, Greene CL, Crider KS, Pollitt RJ. The epidemiology of medium chain acyl-CoA dehydrogenase deficiency: an update. Genet Med. 2006; 8:205–212. PMID: 16617240.
Article7. Shigematsu Y, Hirano S, Hata I, Tanaka Y, Sudo M, Sakura N, et al. Newborn mass screening and selective screening using electrospray tandem mass spectrometry in Japan. J Chromatogr B Analyt Technol Biomed Life Sci. 2002; 776:39–48.
Article8. Yoon HR, Lee KR, Kang S, Lee DH, Yoo HW, Min WK, et al. Screening of newborns and high-risk group of children for inborn metabolic disorders using tandem mass spectrometry in South Korea: a three-year report. Clin Chim Acta. 2005; 354:167–180. PMID: 15748614.
Article9. Purevsuren J, Kobayashi H, Hasegawa Y, Mushimoto Y, Li H, Fukuda S, et al. A novel molecular aspect of Japanese patients with medium-chain acyl-CoA dehydrogenase deficiency (MCADD): c.449-452delCTGA is a common mutation in Japanese patients with MCADD. Mol Genet Metab. 2009; 96:77–79. PMID: 19064330.
Article10. Ensenauer R, Winters JL, Parton PA, Kronn DF, Kim JW, Matern D, et al. Genotypic differences of MCAD deficiency in the Asian population: novel genotype and clinical symptoms preceding newborn screening notification. Genet Med. 2005; 7:339–343. PMID: 15915086.
Article11. Yokoi K, Ito T, Maeda Y, Nakajima Y, Ueta A, Nomura T, et al. Acylcarnitine profiles during carnitine loading and fasting tests in a Japanese patient with medium-chain acyl-CoA dehydrogenase deficiency. Tohoku J Exp Med. 2007; 213:351–359. PMID: 18075239.
Article12. Tajima G, Sakura N, Yofune H, Nishimura Y, Ono H, Hasegawa Y, et al. Enzymatic diagnosis of medium-chain acyl-CoA dehydrogenase deficiency by detecting 2-octenoyl-CoA production using high-performance liquid chromatography: a practical confirmatory test for tandem mass spectrometry newborn screening in Japan. J Chromatogr B Analyt Technol Biomed Life Sci. 2005; 823:122–130.
Article13. Matsubara Y, Kraus JP, Ozasa H, Glassberg R, Finocchiaro G, Ikeda Y, et al. Molecular cloning and nucleotide sequence of cDNA encoding the entire precursor of rat liver medium chain acyl coenzyme A dehydrogenase. J Biol Chem. 1987; 262:10104–10108. PMID: 3611054.
Article14. Waddell L, Wiley V, Carpenter K, Bennetts B, Angel L, Andresen BS, et al. Medium-chain acyl-CoA dehydrogenase deficiency: genotype-biochemical phenotype correlations. Mol Genet Metab. 2006; 87:32–39. PMID: 16291504.
Article15. Van Hove JL, Zhang W, Kahler SG, Roe CR, Chen YT, Terada N, et al. Medium-chain acyl-CoA dehydrogenase (MCAD) deficiency: diagnosis by acylcarnitine analysis in blood. Am J Hum Genet. 1993; 52:958–966. PMID: 8488845.16. Zschocke J, Schulze A, Lindner M, Fiesel S, Olgemöller K, Hoffmann GF, et al. Molecular and functional characterisation of mild MCAD deficiency. Hum Genet. 2001; 108:404–408. PMID: 11409868.
Article17. Nagao M. Frequency of 985A-to-G mutation in medium-chain acyl-CoA dehydrogenase gene among patients with sudden infant death syndrome, Reye syndrome, severe motor and intellectual disabilities and healthy newborns in Japan. Acta Paediatr Jpn. 1996; 38:304–307. PMID: 8840534.
Article18. Tanaka K, Gregersen N, Ribes A, Kim J, Kølvraa S, Winter V, et al. A survey of the newborn populations in Belgium, Germany, Poland, Czech Republic, Hungary, Bulgaria, Spain, Turkey, and Japan for the G985 variant allele with haplotype analysis at the medium chain Acyl-CoA dehydrogenase gene locus: clinical and evolutionary consideration. Pediatr Res. 1997; 41:201–209. PMID: 9029639.19. Zytkovicz TH, Fitzgerald EF, Marsden D, Larson CA, Shih VE, Johnson DM, et al. Tandem mass spectrometric analysis for amino, organic, and fatty acid disorders in newborn dried blood spots: a two-year summary from the New England Newborn Screening Program. Clin Chem. 2001; 47:1945–1955. PMID: 11673361.20. Hsu HW, Zytkovicz TH, Comeau AM, Strauss AW, Marsden D, Shih VE, et al. Spectrum of medium-chain acyl-CoA dehydrogenase deficiency detected by newborn screening. Pediatrics. 2008; 121:e1108–e1114. PMID: 18450854.
Article21. Flanagan SE, Patch AM, Ellard S. Using SIFT and PolyPhen to predict loss-of-function and gain-of-function mutations. Genet Test Mol Biomarkers. 2010; 14:533–537. PMID: 20642364.
Article22. Ng PC, Henikoff S. Accounting for human polymorphisms predicted to affect protein function. Genome Res. 2002; 12:436–446. PMID: 11875032.
Article23. Arnold GL, Saavedra-Matiz CA, Galvin-Parton PA, Erbe R, Devincentis E, Kronn D, et al. Lack of genotype-phenotype correlations and outcome in MCAD deficiency diagnosed by newborn screening in New York State. Mol Genet Metab. 2010; 99:263–268. PMID: 20036593.
Article24. Andresen BS, Bross P, Jensen TG, Winter V, Knudsen I, Kølvraa S, et al. A rare disease-associated mutation in the medium-chain acyl-CoA dehydrogenase (MCAD) gene changes a conserved arginine, previously shown to be functionally essential in short-chain acyl-CoA dehydrogenase (SCAD). Am J Hum Genet. 1993; 53:730–739. PMID: 8102510.
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- A Suspect Case of Medium Chanin Acyl-Coenzyme A Dehydrogenase Deficiency with Iron Deficiency Anemia
- Identification of Novel Compound Heterozygous Mutations in the ACADS Gene of an Asymptomatic Korean Newborn with Short Chain Acyl-CoA Dehydrogenase Deficiency by Tandem Mass Spectrometry
- Compound heterozygous mutations of ACADS gene in newborn with short chain acyl-CoA dehydrogenase deficiency: case report and literatures review
- Organic acidemias in Korea
- Inherited metabolic diseases in the urine organic acid analysis of complex febrile seizure patients
