Ophthalmologic Findings of Boucher-Neuhauser Syndrome
- Affiliations
-
- 1Department of Ophthalmology, College of Medicine, Inje University, Pusan, Korea. maekbak@hanmail.net
- 2Ophthalmology Research Foundation, Inje University, Pusan, Korea.
- 3Department of Neurology, College of Medicine, Inje University, Pusan, Korea.
- KMID: 1084216
- DOI: http://doi.org/10.3341/kjo.2008.22.4.263
Abstract
- To report a case of Boucher-Neuhauser syndrome, which is an autosomal recessive disorder characterized by the triad of spinocerebellar ataxia, chorioretinal dystrophy, and hypogonadotropic hypogonadism. An 18-year-old man was seen for visual problems, which had been diagnosed as retinitis pigmentosa at the age of 12 years. His puberty was delayed. At 16 years of age, the patient experienced progressive deterioration of his balance and gait disturbance. Then he was referred to our clinic because Boucher-Neuhauser syndrome was suspected. He had no specific family history; his visual acuity was 0.04 in both eyes. We observed broad retinal pigment epithelium atrophy and degeneration in both fundi. Both fluorescein and indocyanine green angiography showed choriocapillaris atrophy in the posterior pole area and midperiphery. Macular optical coherence tomography showed thinning of the neurosensory retina. An electroretinographic examination showed no photopic or scotopic responses. The Boucher-Neuhauser syndrome should be included in the differential diagnosis of patients with retinitis pigment epithelium atrophy and degeneration.
MeSH Terms
-
Adolescent
Atrophy
Cerebellum/pathology
Coloring Agents/diagnostic use
Electroretinography
Fluorescein Angiography
Humans
Hypogonadism/*diagnosis/genetics
Indocyanine Green/diagnostic use
Magnetic Resonance Imaging
Male
Photoreceptor Cells, Vertebrate/physiology
Retinal Degeneration/*diagnosis/genetics
Retinal Pigment Epithelium/*pathology
Retinitis Pigmentosa/*diagnosis/genetics/physiopathology
Spinocerebellar Degenerations/*diagnosis/genetics
Syndrome
Tomography, Optical Coherence
Figure
-
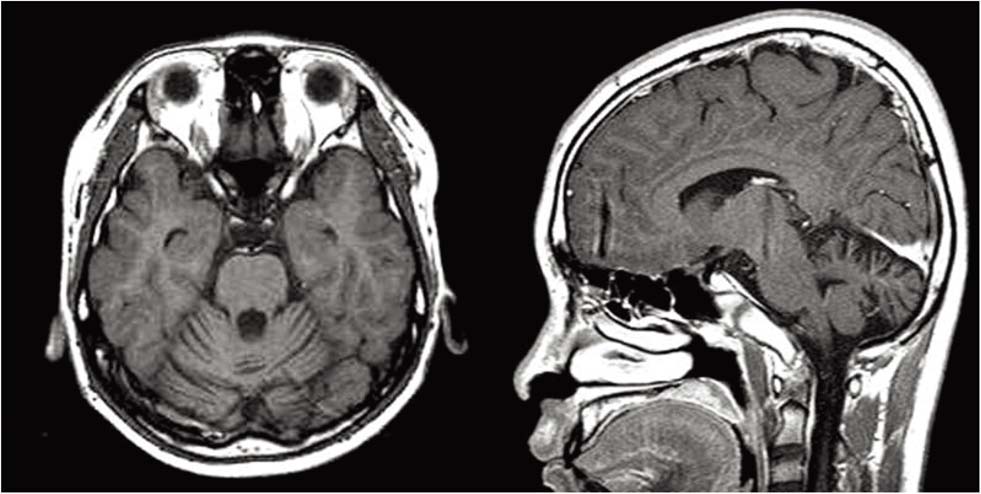
Fig. 1 Brain MRI shows the diffuse atrophy in the cerebellum. Grossly normal pituitary gland.
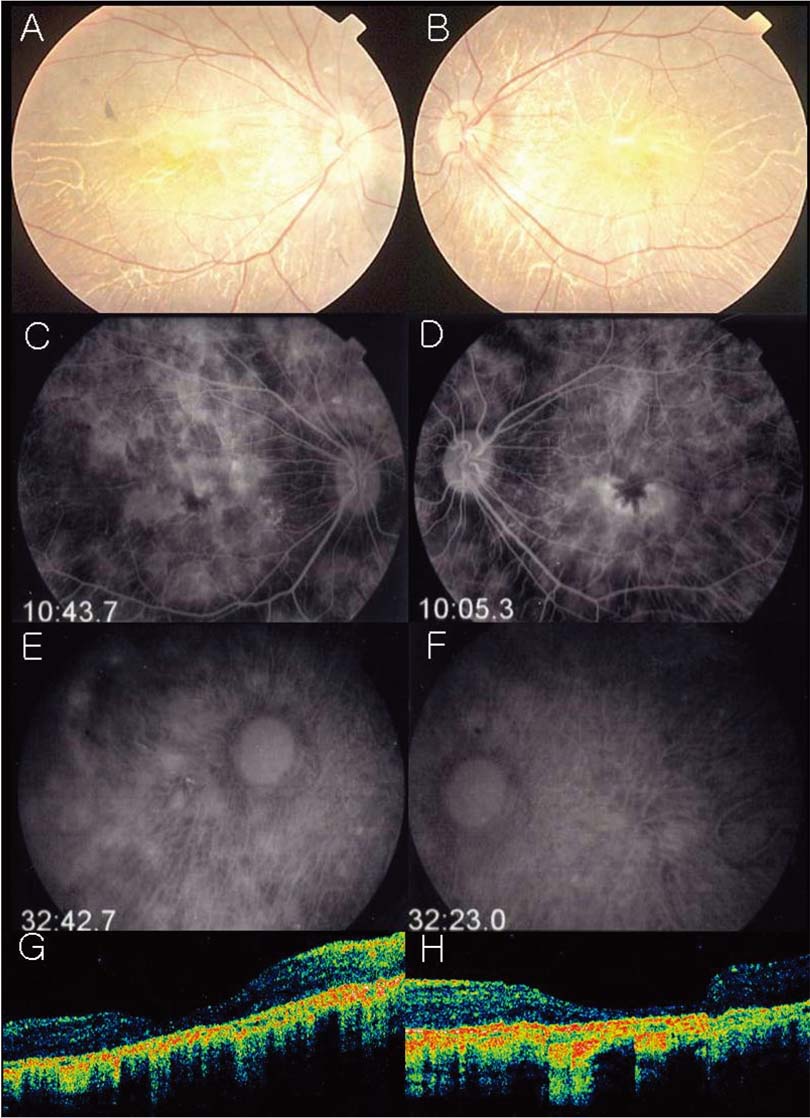
Fig. 2 (A, B) Fundus photograph illustrates a pigmented stippling and marked loss of RPE ; choroidal vessel are easily seen. (C, D) Fluorescein angiogram discloses a profound loss of RPE and choroicapillaris. late arteriovenous-phase fluorescein angiogram shows normal fluorescence of the retinal arteries. Large choroidal vessels can be seen in some areas and the ground-glass fluorescence of the choriocapillaris in other areas where the retinal pigment epithelium and choriocapillaris are more intact. (E, F) Indocyanine green angiography discloses a profound loss of RPE and choroicapillaris. Late arteriovenous-phase fluorescein angiogram shows normal fluorescence of the retinal arteries. Large choroidal vessels can be seen in some areas and the ground-glass fluorescence of the choriocapillaris in other areas where the retinal pigment epithelium and choriocapillaris are more intact. (G, H) OCT showed retinal thining, blunting of the foveal pit, and disappearance of the outer nuclear layer. The foveal center measured 204/ 174 µm.

Fig. 3 Electroretinography shows marked reduced cone and rod response.
Reference
-
1. Boucher BJ, Gibberd FB. Familial ataxia, hypogonadism and retinal degeneration. Acta Neurol Scand. 1969. 45:507–510.2. Neuhäuser G, Opitz JM. Autosomal recessive syndrome of cerebellar ataxia and hypogonadotropic hypogonadism. Clin Genet. 1975. 7:426–434.3. Fok AC, Wong MC, Cheah JS. Syndrome of cerebellar ataxia and hypogonadotrophic hypogonadism: evidence for pituitary gonadotrophin deficiency. J Neurol Neurosurg Psychiatry. 1989. 52:407–409.4. Baronicini A, Franco N, Forabosco A. A new family with chorioretinal dystrophy, spinocerebellar ataxia and hypogonadotropic hypogonadism (Boucher-Neuhauser syndrome). Clin Genet. 1991. 39:274–277.5. Limber ER, Bresnick GH, Lebovitz RM, et al. Spinocerebellar ataxia, hypogonadotropic hypogonadism, and choroidal dystrophny (Boucher-Neuhäuser syndrome). Am J Med Genet. 1994. 53:393–394.6. Traboulsi EI, Maumenee IH, Green WR, et al. Olivopontocerebellar atrophy wirh retinal degeneration. A clinical and ocular histopathologic study. Arch Ophthalmol. 1988. 106:801–806.7. Konigsmark BW, Weiner LP. The olivopontocerebellar atrophies: a review. Medicine (Baltimore). 1970. 49:227–241.8. Drack AV, Traboulsi EI, Maumenee IH. Progression of retinopathy in olivopontocerebellar atrophy with retinal degeneration. Arch Ophthalmol. 1992. 110:712–713.9. François J. Metabolic tapetoretinal degenerations. Surv Ophthalmol. 1982. 26:293–333.10. Kornzweig AL, Bassen FA. Retinitis pigmentosa, acanthrocytosis, and heredodegenerative neuromuscular disease. AMA Arch Ophthalmol. 1957. 58:183–187.11. Salvador F, Garćia-Arumí J, Corcóstegui B, et al. Ophthalmologic findings in a patient with cerebellar ataxia, hypogonadotropic hypogonadism, and chorioretinal dystrophy. Am J ophthalmol. 1995. 120:241–244.
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- The First Korean Family With Boucher-Neuhäuser Syndrome Carrying a Novel Mutation in PNPLA6
- Ocular manifestations in Leigh syndrome
- Vestibular Migraine, How Accurately Can It Be Diagnosed? Prevalence of Preliminary Clinical Diagnosis at a Dizziness Clinic and Prevalence Based on Bárány and Neuhauser Diagnostic Criteria
- Ophthalmologic Features of Lennox-Gastaut Syndrome
- Visual Evoked Potentials in Guillain-Barre Syndrome
