Charcot-Marie-Tooth Disease: Seventeen Causative Genes
- Affiliations
-
- 1Department of Neurology and Ewha Medical Research Center, College of Medicine, Ewha Womans University, Seoul, Korea. bochoi@ewha.ac.kr
- KMID: 1700741
- DOI: http://doi.org/10.3988/jcn.2006.2.2.92
Abstract
- Charcot-Marie-Tooth disease (CMT) is the most common form of inherited motor and sensory neuropathy. Moreover, CMT is a genetically heterogeneous disorder of the peripheral nervous system, with many genes identified as CMT-causative. CMT has two usual classifications: type 1, the demyelinating form (CMT1); and type 2, the axonal form (CMT2). In addition, patients are classified as CMTX if they have an X-linked inheritance pattern and CMT4 if the inheritance pattern is autosomal recessive. A large amount of new information on the genetic causes of CMT has become available, and mutations causing it have been associated with more than 17 different genes and 25 chromosomal loci. Advances in our understanding of the molecular basis of CMT have revealed an enormous diversity in genetic mechanisms, despite a clinical entity that is relatively uniform in presentation. In addition, recent encouraging studies - shown in CMT1A animal models - concerning the therapeutic effects of certain chemicals have been published; these suggest potential therapies for the most common form of CMT, CMT1A. This review focuses on the inherited motor and sensory neuropathy subgroup for which there has been an explosion of new molecular genetic information over the past decade.
Keyword
MeSH Terms
Figure
-

Figure 1 Contemporary portraits of Charcot (A), Marie (B), and Tooth (C). In 1886, Frenchmen Jean Martin Charcot (1825~1893) and Pierre Marie (1853~1940), and Briton Howard Henry Tooth (1856~1925), described hereditary motor and sensory neuropathies for the first time.
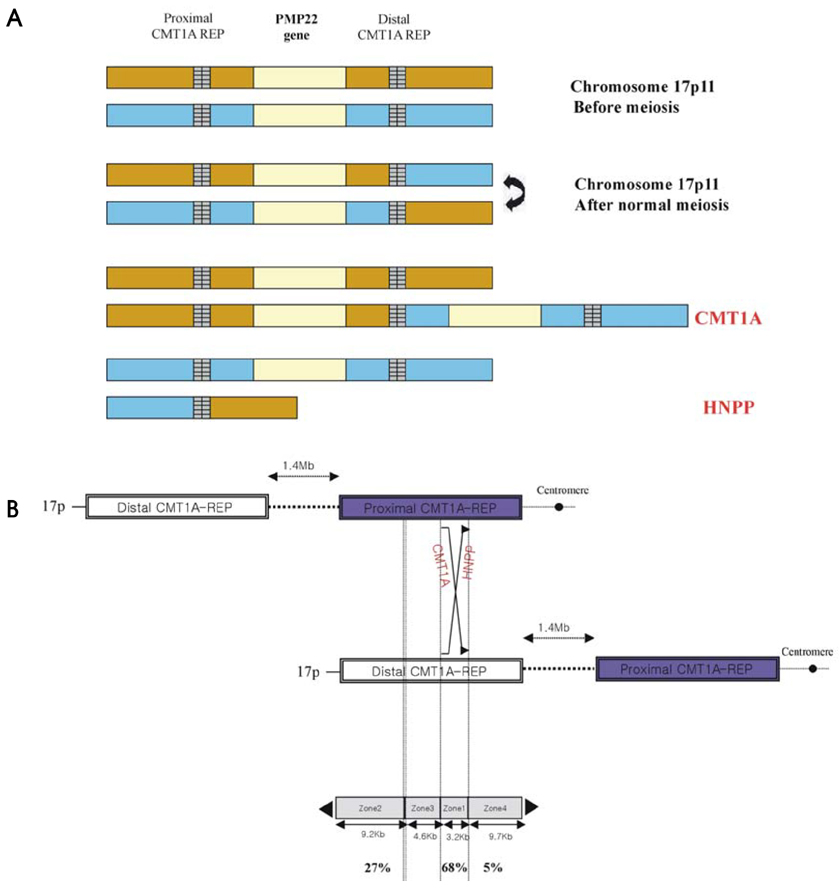
Figure 2 (A) CMT1A duplication and HNPP deletion are reciprocal products of a recombination event (unequal crossing-over) during meiosis, mediated by the flanking repeat elements (CMT1A-REPs). (B) Genomic map of the chromosome 17p11.2-p12. The genomic structure of the CMT1A/HNPP region (telomere-to-centromere orientation). Proximal and distal CMT1A-REPs are shown as vertical boxes.

Figure 3 Genomic structures of GJB1 (A) and EGR2 (B). The EGR2 gene encodes a zinc finger transcription factor that plays a major role in myelination of the peripheral nerves, and the EGR2 gene regulates the expressions of GJB1. Thus, mutations in EGR2 prevent Schwann cell development and lead to the development of demyelinating neuropathy (C).
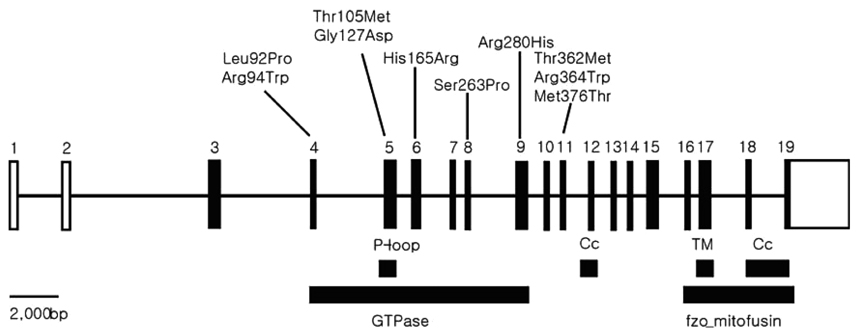
Figure 4 Genomic structure and mutations of MFN2. Solid black boxes and solid white boxes indicate protein coding sequences and untranslated sequences, respectively. P, loop; GTP, binding-site motif; Cc, coiled-coil domain; TM, transmembrane domain; GTPase, GTPase functional domain; fzo mitofusin, fzo mitofusin functional domain.
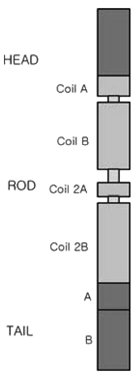
Figure 5 Structure of the NEFL protein, which is one of the most abundant cytoskeletal components of the neuron. The NEFL gene encoding the neurofilament light chain plays an important role in axonal structure, including an extensive fibrous network in the cytoplasm of the neuron.
Cited by 2 articles
-
Exome Sequencing Reveals a Novel PRPS1 Mutation in a Family with CMTX5 without Optic Atrophy
Jin Park, Young Se Hyun, Ye Jin Kim, Soo Hyun Nam, Sung-hee Kim, Young Bin Hong, Jin-Mo Park, Ki Wha Chung, Byung-Ok Choi
J Clin Neurol. 2013;9(4):283-288. doi: 10.3988/jcn.2013.9.4.283.Neuromuscular monitoring of a patient with Charcot-Marie-Tooth disease; which monitoring technique is adequate? - A case report and literature review -
Seung Un Kim, Seora Kim, Ki Tae Jung
Anesth Pain Med. 2024;19(1):54-61. doi: 10.17085/apm.23111.
Reference
-
1. Dyck PJ, Chance P, Lebo R, Carney JA. Dyck PJ, Thomas PK, Griffin JW, Low PA Poduslo JF, editors. Hereditary motor and sensory neuropathies. Peripheral neuropathy. 1993. 3rd edn. Philadelphia: WB Saunders;1094–1136.2. Berger P, Young P, Suter U. Molecular cell biology of Charcot-Marie-Tooth disease. Neurogenetics. 2002. 4:1–15.
Article3. Charcot J, Marie P. Sue une forme particulaire d'atrophie musculaire progressive souvent familial debutant par les pieds et les jamber et atteingnant plus tard les mains. Rev Med. 1886. 6:97–138.4. Tooth H. The peroneal type of progressive muscular atrophy. 1886. London: Lewis.5. Shy ME, Garbern JY, Kamholz J. Hereditary motor and sensory neuropathies: a biological perspective. Lancet Neurol. 2002. 1:110–118.
Article6. Shy ME. Charcot-Marie-Tooth disease: an update. Curr Opin Neurol. 2004. 17:579–585.
Article7. Harding AE, Thomas PK. The clinical features of hereditary motor and sensory neuropathy types I and II. Brain. 1980. 103:259–280.
Article8. Lobsiger CS, Taylor V, Suter U. The early life of a Schwann cell. Biol Chem. 2002. 383:245–253.
Article9. Suter U, Scherer SS. Disease mechanisms in inherited neuropathies. Nat Rev Neurosci. 2003. 4:714–726.
Article10. Sereda MW, Meyer zu Horste G, Suter U, Uzma N, Nave KA. Therapeutic administration of progesterone antagonist in a model of Charcot-Marie-Tooth disease (CMT-1A). Nat Med. 2003. 9:1533–1537.
Article11. Passage E, Norreel JC, Noack-Fraissignes P, Sanguedolce V, Pizant J, Thirion X, et al. Ascorbic acid treatment corrects the phenotype of a mouse model of Charcot-Marie-Tooth disease. Nat Med. 2004. 10:396–401.
Article12. Sahenk Z, Nagaraja HN, McCracken BS, King WM, Freimer ML, Cedarbaum JM, et al. NT-3 promotes nerve regeneration and sensory improvement in CMT1A mouse models and in patients. Neurology. 2005. 65:681–689.
Article13. Martini R. The effect of myelinating Schwann cells on axons. Muscle Nerve. 2001. 24:456–466.
Article14. Huxley C, Passage E, Manson A, Putzu G, Figarella-Branger D, Pellissier JF, et al. Construction of a mouse model of Charcot-Marie-Tooth disease type 1A by pronuclear injection of human YAC DNA. Hum Mol Genet. 1996. 5:563–569.
Article15. Lupski JR. Charcot-Marie-Tooth disease: a gene-dosage effect. Hosp Pract (Minneap). 1997. 32:83–84. 89–91. 94–95.
Article16. Lupski JR, de Oca-Luna RM, Slaugenhaupt S, Pentao L, Guzzetta V, Trask BJ, et al. DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell. 1991. 66:219–232.
Article17. Boerkoel CF, Takashima H, Garcia CA, Olney RK, Johnson J, Berry K, et al. Charcot-Marie-Tooth disease and related neuropathies: mutation distribution and genotype-phenotype correlation. Ann Neurol. 2002. 51:190–201.
Article18. Chance PF, Alderson MK, Leppig KA, Lensch MW, Matsunami N, Smith B, et al. DNA deletion associated with hereditary neuropathy with liability to pressure palsies. Cell. 1993. 72:143–151.
Article19. Young P, Wiebusch H, Strogbauer F, Ringelstein B, Assmann G, Funke H. A novel frameshift mutation in PMP22 accounts for hereditary neuropathy with liability to pressure palsies. Neurology. 1997. 48:450–452.
Article20. Nicholson GA, Valentijn LJ, Cherryson AK, Kennerson ML, Bragg TL, DeKroon RM, et al. A frame shift mutation in the PMP22 gene in hereditary neuropathy with liability to pressure palsies. Nat Genet. 1994. 6:263–266.
Article21. Sereda M, Griffiths I, Puhlhofer A, Stewart H, Rossner MJ, Zimmerman F, et al. A transgenic rat model of Charcot-Marie-Tooth disease. Neuron. 1996. 16:1049–1060.
Article22. Behse F, Buchthal F, Carlsen F, Knappeis GG. Hereditary neuropathy with liability to pressure palsies. Electrophysiological and histopathological aspects. Brain. 1972. 95:777–794.
Article23. Verhagen WI, Gabreels-Festen AA, van Wensen PJ, Joosten EM, Vingerhoets HM, Gabreels FJ, et al. Hereditary neuropathy with liability to pressure palsies: a clinical, electroneurophysiological and morphological study. J Neurol Sci. 1993. 116:176–184.
Article24. Le Guern E, Sturtz F, Gugenheim M, Gouider R, Bonnebouche C, Ravise N, et al. Detection of deletion within 17p11.2 in 7 French families with hereditary neuropathy with liability to pressure palsies (HNPP). Cytogenet Cell Genet. 1994. 65:261–264.
Article25. Mariman FC, Sillekens PT, van Beek-Reinders, van Venrooij WJ. A model for the excision of introns 1 and 2 from adenoviral major late pre-messenger RNAs. J Mol Biology. 1984. 178:47–62.
Article26. Gouider R, LeGuern E, Emile J, Tardieu S, Cabon F, Samid M, et al. Hereditary neuralgic amyotrophy and hereditary neuropathy with liability to pressure palsies: two distinct clinical, electrophysiologic, and genetic entities. Neurology. 1994. 44:2250–2252.
Article27. Pareyson D, Scaioli V, Taroni F, Botti S, Lorenzetti D, Solari A, et al. Phenotypic heterogeneity in hereditary neuropathy with liability to pressure palsies associated with chromosome 17p11.2-12 deletion. Neurology. 1996. 46:1133–1137.
Article28. Kim SM, Chung KW, Choi BO, Yoon ES, Choi JY, Park KD, et al. Hereditary neuropathy with liability to pressure palsies (HNPP) patients of Korean ancestry with chromosome 17p11.2-p12 deletion. Exp Mol Med. 2004. 36:28–35.
Article29. Roa BB, Garcia CA, Suter U, Kulpa DA, Wise CA, Mueller J, et al. Charcot-Marie-Tooth disease type 1A. Association with a spontaneous point mutation in the PMP22 gene. N Engl J Med. 1993. 329:96–101.
Article30. Roa BB, Dyck PJ, Marks HG, Chance PF, Lupski JR. Déjérine-Sottas syndrome associated with point mutation in the peripheral myelin protein 22 (PMP22) gene. Nat Genet. 1993. 5:269–273.
Article31. Choi BO, Chung KW, Park KD, Choi KG, Kim SM, Kim Y, et al. Charcot-Marie-Tooth type 1A patient with a novel frame shift mutation (Ala106fs) in the PMP22 gene. J Korean Neurol Assoc. 2004. 22:673–676.32. Timmerman V, De Jonghe P, Ceuterick C, De Vriendt E, Lofgren A, Nelis E, et al. Novel missense mutation in the early growth response 2 gene associated with Dejerine-Sottas syndrome phenotype. Neurology. 1999. 52:1827–1832.
Article33. Warner LE, Mancias P, Butler IJ, McDonald CM, Keppen L, Koob KG, et al. Mutations in the early growth response 2 (EGR2) gene are associated with hereditary myelinopathies. Nat Genet. 1998. 18:382–384.
Article34. Topilko P, Schneider-Maunoury S, Levi G, Baron-Van Evercooren A, Chennoufi AB, Seitanidou T, et al. Krox-20 controls myelination in the peripheral nervous system. Nature. 1994. 371:796–799.
Article35. Nagarajan R, Svaren J, Le N, Araki T, Watson M, Milbrandt J. EGR2 mutations in inherited neuropathies dominant-negatively inhibit myelin gene expression. Neuron. 2001. 30:355–368.
Article36. Chung KW, Sunwoo IN, Kim SM, Park KD, Kim WK, Kim TS, et al. Two missense mutations of EGR2 R359W and GJB1 V136A in a Charcot-Marie-Tooth disease family. Neurogenetics. 2005. 6:159–163.
Article37. Musso M, Balestra P, Bellone E, Cassandrini D, Di Maria E, Doria LL, et al. The D355V mutation decreases EGR2 binding to an element within the Cx32 promoter. Neurobiol Dis. 2001. 8:700–706.
Article38. Warner LE, Svaren J, Milbrandt J, Lupski JR. Functional consequences of mutations in the early growth response 2 gene (EGR2) correlate with severity of human myelinopathies. Hum Mol Genet. 1999. 8:1245–1251.
Article39. Tang X, Fenton MJ, Amar S. Identification and functional characterization of a novel binding site on TNF-alpha promoter. Proc Natl Acad Sci U S A. 2003. 100:4096–4101.
Article40. Moriwaki Y, Begum NA, Kobayashi M, Matsumoto M, Toyoshima K, Seya T. Mycobacterium bovis Bacillus Calmette-Guerin and its cell wall complex induce a novel lysosomal membrane protein, SIMPLE, that bridges the missing link between lipopolysaccharide and p53-inducible gene, LITAF (PIG7), and estrogen-inducible gene, EET-1. J Biol Chem. 2001. 276:23065–23076.
Article41. Guilbot A, Williams A, Ravise N, Verny C, Brice A, Sherman DL, et al. A mutation in periaxin is responsible for CMT4F, an autosomal recessive form of Charcot-Marie-Tooth disease. Hum Mol Genet. 2001. 10:415–421.
Article42. Gillespie CS, Sherman DL, Blair GE, Brophy PJ. Periaxin, a novel protein of myelinating Schwann cells with a possible role in axonal ensheathment. Neuron. 1994. 12:497–508.
Article43. Sherman DL, Brophy PJ. A tripartite nuclear localization signal in the PDZ-domain protein L-periaxin. J Biol Chem. 2000. 275:4537–4540.
Article44. Scherer SS, Xu YT, Bannerman PG, Sherman DL, Brophy PJ. Periaxin expression in myelinating Schwann cells: modulation by axon-glial interactions and polarized localization during development. Development. 1995. 121:4265–4273.
Article45. Kijima K, Numakura C, Shirahata E, Sawaishi Y, Shimohata M, Igarashi S, et al. Periaxin mutation causes early-onset but slow-progressive Charcot-Marie-Tooth disease. J Hum Genet. 2004. 49:376–379.
Article46. Bolino A, Muglia M, Conforti FL, LeGuern E, Salih MA, Georgiou DM, et al. Charcot-Marie-Tooth type 4B is caused by mutations in the gene encoding myotubularinrelated protein-2. Nat Genet. 2000. 25:17–19.
Article47. Laporte J, Blondeau F, Buj-Bello A, Mandel JL. The myotubularin family: from genetic disease to phosphoinositide metabolism. Trends Genet. 2001. 17:221–228.
Article48. Gambardella A, Bono F, Muglia M, Valentino P, Quattrone A. Autosomal recessive hereditary motor and sensory neuropathy with focally folded myelin sheaths (CMT4B). Ann NY Acad Sci. 1999. 883:47–55.
Article49. Senderek J, Bergmann C, Weber S, Ketelsen UP, Schorle H, Rudnik-Schoneborn S, et al. Mutation of the SBF2 gene, encoding a novel member of the myotubularin family, in Charcot-Marie-Tooth neuropathy type 4B2/11p15. Hum Mol Genet. 2003. 12:349–356.
Article50. Züchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, Rochelle J, Dadali EL, et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet. 2004. 36:449–451.
Article51. Kijima K, Numakura C, Izumino H, Umetsu K, Nezu A, Shiiki T, et al. Mitochondrial GTPase mitofusin 2 mutation in Charcot-Marie-Tooth neuropathy type 2A. Hum Genet. 2005. 116:23–27.
Article52. Lawson VH, Graham BV, Flanigan KM. Clinical and electrophysiologic features of CMT2A with mutations in the mitofusin 2 gene. Neurology. 2005. 65:197–204.
Article53. Rojo M, Legros F, Chateau D, Lombes A. Membrane topology and mitochondrial targeting of mitofusins, ubiquitous mammalian homologs of the transmembrane GTPase Fzo. J Cell Sci. 2002. 115:1663–1674.
Article54. Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003. 160:189–200.
Article55. Koshiba T, Detmer SA, Kaiser JT, Chen H, McCaffery JM, Chan DC. Structural basis of mitochondrial tethering by mitofusin complexes. Science. 2004. 305:858–862.
Article56. Chen H, Chomyn A, Chan DC. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem. 2005. 280:26185–26192.
Article57. Honda S, Aihara T, Hontani M, Okubo K, Hirose S. Mutational analysis of action of mitochondrial fusion factor mitofusin-2. J Cell Sci. 2005. 118:3153–3161.
Article58. Szigeti K, Garcia CA, Lupski JR. Charcot-Marie-Tooth disease and related hereditary polyneuropathies: molecular diagnostics determine aspects of medical management. Genet Med. 2006. 8:86–92.
Article59. Züchner S, De Jonghe P, Jordanova A, Claeys KG, Guergueltcheva V, Cherninkova S, et al. Axonal neuropathy with optic atrophy is caused by mutations in mitofusin 2. Ann Neurol. 2006. 59:276–281.
Article60. Choi BO, Kim SB, Park KD, Choi KG, Oh J, Suh BC, et al. Clinical and genetic characteristics in patients of Charcot-Marie-Tooth type 2A with mitofusin 2 (MFN2) mutations. J Korean Neurol Assoc. 2006. 24:131–140.61. Zhao C, Takita J, Tanaka Y, Setou M, Nakagawa T, Takeda S, et al. Charcot-Marie-Tooth disease type 2A caused by mutation in a microtubule motor KIF1B beta. Cell. 2001. 105:587–597.
Article62. Bissar-Tadmouri N, Nelis E, Züchner S, Parman Y, Deymeer F, Serdaroglu P, et al. Absence of KIF1B mutation in a large Turkish CMT2A family suggests involvement of a second gene. Neurology. 2004. 62:1522–1525.
Article63. Muglia M, Zappia M, Timmerman V, Valentino P, Gabriele AL, Conforti FL, et al. Clinical and genetic study of a large Charcot-Marie-Tooth type 2A family from southern Italy. Neurology. 2001. 56:100–103.
Article64. Verhoeven K, De Jonghe P, Coen K, Verpoorten N, Auer-Grumbach M, Kwon JM, et al. Mutations in the small GTP-ase late endosomal protein RAB7 cause Charcot-Marie-Tooth type 2B neuropathy. Am J Hum Genet. 2003. 72:722–727.
Article65. Jordens I, Fernandez-Borja M, Marsman M, Dusseljee S, Janssen L, Calafat J, et al. The Rab7 effector protein RILP controls lysosomal transport by inducing the recruitment of dynein-dynactin motors. Curr Biol. 2001. 11:1680–1685.
Article66. Irobi J, Van Impe K, Seeman P, Jordanova A, Dierick I, Verpoorten N, et al. Hot-spot residue in small heat-shock protein 22 causes distal motor neuropathy. Nat Genet. 2004. 36:597–601.
Article67. Benndorf R, Welsh MJ. Shocking degeneration. Nat Genet. 2004. 36:547–548.
Article68. Evgrafov OV, Mersiyanova I, Irobi J, Van Den Bosch L, Dierick I, Leung CL, et al. Mutant small heat-shock protein 27 causes axonal Charcot-Marie-Tooth disease and distal hereditary motor neuropathy. Nat Genet. 2004. 36:602–606.
Article69. Sun X, Fontaine JM, Rest JS, Shelden FA, Welsh MJ, Benndorf R. Interaction of human HSP22 (HSPB8) with other small heat shock proteins. J Biol Chem. 2004. 279:2394–2402.
Article70. Benndorf R, Sun X, Gilmont RR, Biederman KJ, Molloy MP, Goodmurphy CW, et al. HSP22, a new member of the small heat shock protein superfamily, interacts with mimic of phosphorylated HSP27 ((3D)HSP27). J Biol Chem. 2001. 276:26753–26761.
Article71. Antonellis A, Ellsworth RE, Sambuughin N, Puls I, Abel A, Lee-Lin SQ, et al. Glycyl tRNA synthetase mutations in Charcot-Marie-Tooth disease type 2D and distal spinal muscular atrophy type V. Am J Hum Genet. 2003. 72:1293–1299.
Article72. Warner LE, Hilz MJ, Appel SH, Killian JM, Kolodry EH, Karpati G, et al. Clinical phenotypes of different MPZ (P0) mutations may include Charcot-Marie-Tooth type IB, Déjérine-Sottas, and congenital hypomyelination. Neuron. 1996. 17:451–460.
Article73. Marrosu MG, Vaccargiu S, Marrosu G, Vannelli A, Cianchetti C, Muntoni F. Charcot-Marie-Tooth disease type 2 associated with mutation of the myelin protein zero gene. Neurology. 1998. 50:1397–1401.
Article74. Shy ME, Jani A, Krajewski K, Grandis M, Lewis RA, Li J, et al. Phenotypic clustering in MPZ mutations. Brain. 2004. 127:371–384.
Article75. Hattori N, Yamamoto M, Yoshihara T, Koike H, Nakagawa M, Yoshikawa H, et al. Demyelinating and axonal features of Charcot-Marie-Tooth disease with mutations of myelinrelated proteins (PMP22, MPZ and Cx32): a clinicopathological study of 205 Japanese patients. Brain. 2003. 126:134–151.
Article76. Lemke G, Axel R. Isolation and sequence of a cDNA encoding the major structural protein of peripheral myelin. Cell. 1985. 40:501–508.
Article77. Xu W, Manichella D, Jiang H, Vallat Jm, Lilien J, Baron P, et al. Absence of P0 leads to the dysregulation of myelin gene expression and myelin morphogenesis. J Neurosci Res. 2000. 60:714–724.
Article78. Wong MH, Filbin MT. The cytoplasmic domain of the myelin P0 protein influences the adhesive interactions of its extracellular domain. J Cell Biol. 1994. 126:1089–1097.
Article79. Carter J, Gragerov A, Konvicka K, Elder G, Weinstein H, Lazzarini RA. Neurofilament (NF) assembly; divergent characteristics of human and rodent NF-L subunits. J Biol Chem. 1998. 273:5101–5108.
Article80. Fabrizi GM, Cavallaro T, Angiari C, Bertolasi L, Cabrini I, Ferrarini M, et al. Giant axon and neurofilament accumulation in Charcot-Marie-Tooth disease type 2E. Neurology. 2004. 62:1429–1431.
Article81. De Jonghe P, Mersivanova I, Nelis E, Del Favero J, Martin JJ, van Broeckhoven C, et al. Further evidence that neurofilament light chain gene mutations can cause Charcot-Marie-Tooth disease type 2E. Ann Neurol. 2001. 49:245–249.
Article82. Zhu Q, Couillard-Despres S, Julien JP. Delayed maturation of regenerating myelinated axons in mice lacking neurofilaments. Exp Neurol. 1997. 148:299–316.
Article83. Kriz J, Zhu Q, Julien JP, Padjen AL. Electrophysiological properties of axons in mice lacking neurofilament subunit genes: disparity between conduction velocity and axon diameter in absence of NF-H. Brain Res. 2000. 885:32–44.
Article84. Jordanova A, De Jonghe P, Boerkoel CF, Takashima H, De Vriendt E, Ceuterick C, et al. Mutations in the neurofilament light chain gene (NEFL) cause early onset severe Charcot-Marie-Tooth disease type 1A. Brain. 2003. 126:590–597.
Article85. Choi BO, Lee MS, Shin SH, Hwang JH, Choi KG, Kim WK, et al. Mutational analysis of PMP22, MPZ, GJB1, EGR2 and NEFL in Korean Charcot-Marie-Tooth neuropathy patients. Hum Mutat. 2004. 24:185–186.86. Herringham WP. Muscular atrophy of the peroneal type affecting many members of a family. Brain. 1889. 11:230–236.
Article87. Bergoffen J, Scherer SS, Wang S, Scott MO, Bone LJ, Paul DL, et al. Connexin mutations in X-linked Charcot-Marie-Tooth disease. Science. 1993. 262:2039–2042.
Article88. Birouk N, LeGuern E, Maisonobe T, Rouger H, Gouider R, Tardieu S, et al. X-linked Charcot-Marie-Tooth disease with connexin 32 mutations: clinical and electrophysiologic study. Neurology. 1998. 50:1074–1082.
Article89. Hahn AF, Bolton CF, White CM, Brown WF, Tuuha SE, Tan CC, et al. Genotype/phenotype correlations in Xlinked dominant Charcot-Marie-Tooth disease. Ann N Y Acad Sci. 1999. 883:366–382.
Article90. Fischbeck KH, ar-Rushdi N, Pericak-Vance M, Rozear M, Roses AD, Fryns JP. X-linked neuropathy: gene localization with DNA probes. Ann Neurol. 1986. 20:527–532.
Article91. Vital A, Ferrer X, Lagueny A, Vandenberghe A, Latour P, Goizet C, et al. Histopathological features of X-linked Charcot-Marie-Tooth disease in 8 patients from 6 families with different connexin32 mutations. J Peripher Nerv Syst. 2001. 6:79–84.
Article92. Nicholson SM, Gomes D, de Nechaud B, Bruzzone R. Altered gene expression in Schwann cells of connexin32 knockout animals. J Neurosci Res. 2001. 66:23–36.
Article93. Evans WH, Martin PE. Gap junctions: structure and function. Mol Membr Biol. 2002. 19:121–136.94. Kumar NM, Gilula NB. Cloning and characterization of human and rat liver cDNAs coding for a gap junction protein. J Cell Biol. 1986. 103:767–776.
Article95. Scherer SS, Deschenes SM, Xu YT, Grinspan JB, Fischbeck KH, Paul DL. Connexin32 is a myelin-related protein in the PNS and CNS. J Neurosci. 1995. 15:8281–8294.
Article96. Rozear MP, Pericak-Vance MA, Fischbeck K, Stajich JM, Gaskell PC Jr, Krendel DA, et al. Hereditary motor and sensory neuropathy, X-linked: a half century follow-up. Neurology. 1987. 37:1460–1465.
Article97. Hahn AF, Brown WF, Koopman WJ, Feasby TE. X-linked dominant hereditary motor and sensory neuropathy. Brain. 1990. 113:1511–1525.
Article98. Bort S, Nelis E, Timmerman V, Sevilla T, Cruz-Martinez A, Martinez F, et al. Mutational analysis of the MPZ, PMP22 and Cx32 genes in patients of Spanish ancestry with Charcot-Marie-Tooth disease and hereditary neuropathy with liability to pressure palsies. Hum Genet. 1997. 99:746–754.
Article99. Silander K, Meretoja P, Juvonen V, Ignatius J, Pihko H, Saarinen A, et al. Spectrum of mutations in Finnish patients with Charcot-Marie-Tooth disease and related neuropathies. Hum Mutat. 1998. 12:59–68.
Article100. Mersiyanova IV, Ismailov SM, Polyakov AV, Dadali EL, Fedotov VP, Nelis E, et al. Screening for mutations in the peripheral myelin genes PMP22, MPZ and Cx32 (GJB1) in Russian Charcot-Marie-Tooth neuropathy patients. Hum Mutat. 2000. 15:340–347.
Article101. Mostacciuolo ML, Righetti E, Zortea M, Bosello V, Schiavon F, Vallo L, et al. Charcot-Marie-Tooth disease type 1 and related demyelinating neuropathies: mutation analysis in a large cohort of Italian families. Hum Mutat. 2001. 18:32–41.
Article102. Huehne K, Benes V, Thiel C, Kraus C, Kress W, Hoeltzenbein M, et al. Novel mutations in the Charcot-Marie-Tooth disease genes PMP22, MPZ, and GJB1. Hum Mutat. 2003. 21:100.
Article103. Yoshihara T, Yamamoto M, Doyu M, Mis KI, Hattori N, Hasegawa Y, et al. Mutations in the peripheral myelin protein zero and connexin32 genes detected by nonisotopic RNase cleavage assay and their phenotypes in Japanese patients with Charcot-Marie-Tooth disease. Hum Mutat. 2000. 16:177–178.
Article104. Numakura C, Lin C, Ikegami T, Guldberg P, Hayasaka K. Molecular analysis in Japanese patients with Charcot-Marie-Tooth disease: DGGE analysis for PMP22, MPZ, and Cx32/GJB1 mutations. Hum Mutat. 2002. 20:392–398.
Article105. Nelis E, Erdem S, Van Den Bergh PY, Belpaire-Dethiou MC, Ceuterick C, Van Gerwen V, et al. Mutations in GDAP1: autosomal recessive CMT with demyelination and axonopathy. Neurology. 2002. 59:1865–1872.
Article106. Baxter RV, Ben-Othmane K, Rochelle JM, Stajich JE, Hulette C, Dew-Knight S, et al. Ganglioside-induced differentiation-associated protein-1 is mutant in Charcot-Marie-Tooth disease type 4A/8q21. Nat Genet. 2002. 30:21–22.
Article107. Cuesta A, Pedrola L, Sevilla T, Garcia-Planells J, Chumillas MJ, Mayordomo F, et al. The gene encoding ganglioside-induced differentiation-associated protein 1 is mutated in axonal Charcot-Marie-Tooth type 4A disease. Nat Genet. 2002. 30:22–25.
Article108. Asbury AK, Gale MK, Cox SC, Baringer JR, Berg BO. Giant axonal neuropathy - a unique case with segmental neurofilamentous masses. Acta Neuropathol (Berl). 1972. 20:237–247.
Article109. Bomont P, Cavalier L, Blondeau F, Ben Hamida C, Belal S, Tazir M, et al. The gene encoding gigaxonin, a new member of the cytoskeletal BTB/kelch repeat family, is mutated in giant axonal neuropathy. Nat Genet. 2000. 26:370–374.
Article110. Senderek J, Bergmann C, Stendel C, Kirfel J, Verpoorten N, De Jonghe P, et al. Mutations in a gene encoding a novel SH3/TPR domain protein cause autosomal recessive Charcot-Marie-Tooth type 4C neuropathy. Am J Hum Genet. 2003. 73:1106–1119.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Clinical and genetic aspects of Charcot-Marie-Tooth disease subtypes
- A novel p.Leu699Pro mutation in MFN2 gene causes Charcot-Marie-Tooth disease type 2A
- Operative Treatment of Charcot-Marie-Tooth Disease
- A Case Report of Neuronal Type of Charcot-Marie-Tooth Disease
- DNA diagnostic testing in hereditary motor and sensory neuropathies
